超支化镍系催化剂构效关系及催化乙烯齐聚机理
2020-04-11张娜马立莉陈丽铎李翠勤王俊
张娜,马立莉,陈丽铎,李翠勤,王俊
(1东北石油大学化学化工学院,黑龙江大庆163318;2中国石油化工研究院大庆化工中心,黑龙江大庆163312)
线形α-烯烃是一类重要的有机化工原料和中间体,除可用于合成洗涤剂、润滑剂和增塑剂外,还可用于合成高性能聚烯烃材料[1]。目前工业上生产α-烯烃的主要方法为乙烯齐聚法,其技术核心为催化剂[2-5];后过渡金属铁、钴、镍系催化剂由于具有催化活性高、产物选择性好的优点,近几年在乙烯齐聚领域得到了广泛的应用[6-9]。与铁系和钴系催化剂相比,镍系催化剂因具有较好的热稳定性,且可以通过调节配体结构改变其催化活性和产物选择性等优点而备受关注,从而得到了迅速发展[10]。许多科学家对新型骨架配体的设计合成和催化剂结构与催化性能之间的构效关系进行了深入研究[11-12]。然而,催化过程中所形成活性中心的性质不稳定,并且很难将其从催化体系中分离,这对活性中心性质以及催化乙烯齐聚机理的研究颇有难度。Chandran等[13]采用UV-vis对水杨醛亚胺钴系催化剂催化乙烯齐聚活性中心的形成过程进行了检测,结果表明,催化剂经助催化剂活化后,会在金属中心形成空轨道,乙烯的插入会使活性中心的稳定性增强;朱峰[14]采用电子自旋共振(ESR)技术检测了催化剂SNS-Cr 在催化乙烯齐聚体系中活性中心的形成和变化,提出该催化剂催化乙烯齐聚的反应机理,在反应过程中,Cr3+在MAO作用下形成Cr+,Cr+即为催化活性中心,在催化过程中,铬金属中心的价态在一价和三价之间转变,当提高Al/Cr物质的量之比时,Cr+的电子自旋共振(ESR)信号强度增加,表明活性中心数量增加,催化活性升高。
多支型金属催化剂具有精确的分子结构和良好的单分散性,使其具备同类催化剂没有的催化活性高、热稳定性强以及产物选择性好等优点。与线形α-烯烃相比,多支型金属催化剂分子尺寸较大,可以通过沉淀和过滤的方法将其从反应混合物中分离出来,因此该类催化剂又具备了非均相催化剂的特点。此外,还可以通过调节多支型金属催化剂的结构、尺寸、形状、官能度及溶解性来实现其催化性能的调控[15]。近年来,本文作者课题组合成了系列以超支化大分子为骨架的多支型镍系催化剂,并用于催化乙烯齐聚反应,发现该类镍系催化剂具有较好的催化乙烯齐聚性能,同时催化剂端基和分子空穴处烷基链长度直接影响催化剂的催化活性[16-18]。在以上研究基础上,本文设计合成了系列具有不同空间位阻的超支化水杨醛亚胺配体,与镍配位后得到相应的配合物,并对其催化乙烯齐聚性能进行研究,进而考察配体空间位阻对催化活性及产物分布的影响。
1 实验部分
1.1 试剂及仪器
3-甲基水杨醛、3-叔丁基水杨醛,分析纯,上海梯希爱化学品有限公司;3-苯基水杨醛,分析纯,上海药明康德公司;甲苯、甲基环己烷和环己烷,分析纯,天津科密欧试剂厂,使用前经金属钠回流处理;MAO(质量分数为10%的甲苯溶液)、三乙基铝[Al(Et)3,质量分数为10%的甲苯溶液],Aladdin 公司;NiCl2·6H2O,分析纯,天津市基准化学试剂有限公司;甲醇,分析纯,天津市科密欧化学试剂有限公司;乙醚,分析纯,天津东方化工厂;1.0G超支化大分子,实验室自制[19]。
Vector 22 型傅里叶变换红外光谱仪(瑞士Bruker 公司);micr OTOF-Q II 型电喷雾电离质谱仪(ESI-MS)(美国Bruker 公司);INOV-400MHz型核磁共振仪(美国varian 公司);UV-1700 PharmaSpec 型紫外-可见分光光度计(深圳市科美嘉仪器设备有限公司);Agilent 8800型电感耦合等离子体质谱仪(美国安捷伦公司);GC-9720 型气相色谱仪(浙江福立分析仪器有限公司)。
1.2 超支化水杨醛亚胺镍系催化剂的合成
1.2.1 配体的合成
氮气环境下,将0.01mol 的1.0G 超支化大分子、3.0g 无水硫酸钠和30mL 甲醇依次加入100mL三口瓶中,升温至65℃,回流搅拌15min使其充分溶解,然后缓慢滴加0.04mol的3-取代水杨醛;回流反应12h 后过滤除去硫酸钠固体,滤液于-30℃下静置24h,析出黄色油状液体,分离收集到的油状液体用冷甲醇洗涤3次,减压蒸馏除去溶剂,得到黄色油状液体,即为超支化水杨醛亚胺配体Ligand-1、Ligand-2和Ligand-3,产率分别为65%、69%和64%,合成路线及配体的结构如图1所示。
1.2.2 镍系催化剂的合成
将0.01mol 配体加入到Schlenk 瓶中,将0.01mol 的NiCl2·6H2O 加入到单口瓶中,分别加入10mL 甲醇,得到黄色溶液A 和绿色溶液B,氮气条件下,将溶液B缓慢滴加到溶液A中,25℃下搅拌24h;向反应物中加入50mL 乙醚,析出绿色固体,除去溶剂,固体用乙醚洗涤3 次,过滤、干燥,得到绿色固体粉末,即为超支化镍系催化剂Ni-1、Ni-2和Ni-3,产率分别为75%、77%和78%。元素分析Ni-1(C38H57N5O4Ni):Ni,9.21%,标准,8.36%;Ni-2(C44H69N5O4Ni):Ni,8.06%,标准,7.47%;Ni-3(C48H61N5O4Ni):Ni,7.95%,标准,7.11%。合成路线及镍系催化剂的结构如图1所示。
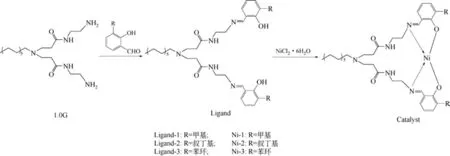
图1 超支化水杨醛亚胺镍系催化剂的合成路线
1.3 乙烯齐聚反应
乙烯齐聚反应在250mL 带磁力搅拌的不锈钢反应釜中进行,反应釜经加热抽真空后,用乙烯气体置换3 次。充入乙烯至常压,依次加入40mL 溶剂和一定量的助催化剂,搅拌5min 后迅速加入10mL 浓度为0.7µmol/mL 的催化剂溶液,充入乙烯至设定压力,在指定温度下进行齐聚反应。持续反应30min,降温、卸压,用质量分数为10%的酸化乙醇终止反应。采用GC-9720 型气相色谱仪分析乙烯齐聚产物,并计算催化剂的催化活性。
2 结果与讨论
2.1 红外光谱分析
采用瑞士Bruker 公司Vector 22 型傅里叶变换红外光谱仪对合成的系列新型超支化配体及其镍系催化剂进行了红外光谱表征,结果如图2。从超支化水杨醛亚胺配体的红外光谱图可以看出,3421cm-1处为的伸缩振动吸收峰;2924cm-1处为及系列的伸缩振动吸收峰;1456cm-1处为苯环骨架的特征吸收峰;747cm-1处为苯环上C H 的面外弯曲振动吸收峰;1635cm-1处为的伸缩振动吸收峰,表明1.0G超支化大分子与系列3-取代水杨醛发生了席夫碱反应。超支化水杨醛亚胺配体与金属镍配位后,催化剂的伸缩振动吸收峰向低位移方向移动,出现在1629cm-1处,且强度减弱。
2.2 核磁共振氢谱分析

图2 超支化水杨醛亚胺配体及其镍系催化剂的红外光谱
采用美国Varian 公司INOV-400MHz 型核磁共振仪对合成的系列超支化水杨醛亚胺配体的核磁共振氢谱进行表征,结果如图3。以超支化水杨醛亚胺配体Ligand-3 为例,在化学位移0.890 处出现分子端基甲基上氢质子的特征峰;在化学位移1.279处出现分子长链烷基中系列亚甲基上氢质子的特征峰;在化学位移2.326和2.634处分别出现与叔胺相连的亚甲基上氢质子的特征峰;在化学位移2.228处出现与羰基相连的亚甲基上氢质子的特征峰;在化学位移6.933 处出现仲胺上氢质子的特征峰;与仲胺和叔胺相连的亚甲基上氢质子的特征峰分别出现在3.372和3.607处;苯环上氢质子的特征峰出现在7.086~7.539处;羟基上氢质子的吸收峰出现在5.290 处;此外,1.0G 超支化大分子与邻苯基水杨醛发生反应生成的席夫碱()上氢质子的吸收峰出现在8.311处。
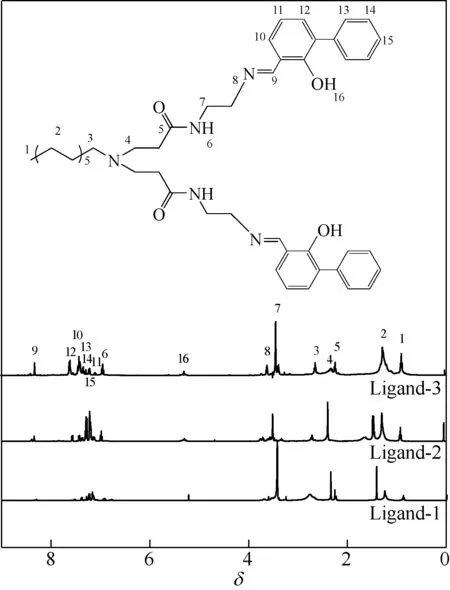
图3 超支化水杨醛亚胺配体的核磁共振氢谱
2.3 紫外光谱分析
采用UV-1700 PharmaSpec型紫外-可见分光光度计对合成的系列超支化水杨醛亚胺配体及其镍系催化剂的紫外可见光光谱进行表征,结果如图4。从超支化水杨醛亚胺配体的紫外-可见光光谱图可以看出,218nm 处出现的n→π*跃迁的R 带;252nm 处出现苯环π→π*跃迁的B 带;329nm 处出现的n→π*跃迁的R带。与超支化水杨醛亚胺配体紫外可见光光谱图相比,镍系催化剂中的n→π*跃迁的R带、苯环π→π*跃迁的B带以及C的n→π*跃迁的R 带均发生蓝移,分别出现在紫外-可见光光谱图中215nm、244nm和325nm处;此外,镍系催化剂紫外谱图中398nm处还出现金属镍d→d*跃迁的吸收谱带,表明超支化水杨醛亚胺配体与金属镍发生了配位反应。
2.4 质谱分析
采用Bruker公司micr OTOF-Q II型电喷雾电离质谱仪对合成的系列超支化水杨醛亚胺镍系催化剂的质谱进行表征,结果如图5。3 种超支化水杨醛亚胺镍系催化剂的准分子离子峰在其质谱中均可以从图5中观察到,其中催化剂Ni-1的准分子离子峰[M+1]+出现在m/z=707.2 处;催化剂Ni-2 的准分子离子峰[M+1]+出现在m/z=791.6 处;催化剂Ni-3 的准分子离子峰[M-1]+出现在m/z=830.4处。
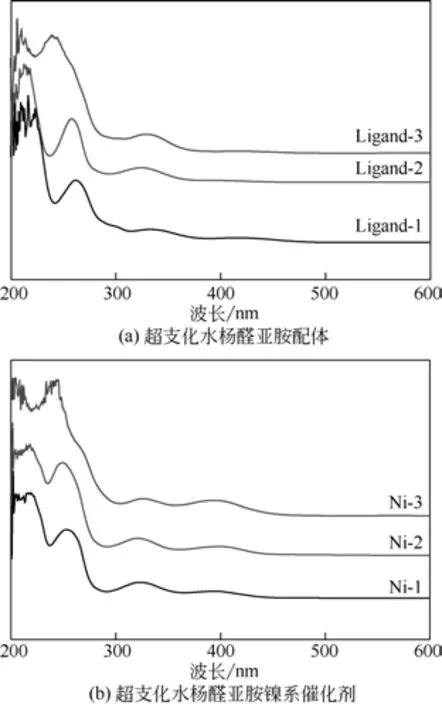
图4 超支化水杨醛亚胺配体及其镍系催化剂的紫外-可见光光谱图
2.5 配体空间位阻对催化乙烯齐聚性能的影响
配体空间位阻对超支化水杨醛亚胺镍系催化剂催化乙烯齐聚活性和高碳烯烃选择性的影响结果见表1。由表1 可以看出,通过改变超支化水杨醛亚胺配体的空间位阻可以有效地提高镍系催化剂的催化性能。配体苯环上取代基的空间位阻越大,催化剂的活性越高,催化活性从高到低的顺序为:Ni-3>Ni-2>Ni-1。这可能是因为,当取代基的空间位阻较大时,能够更有效地屏蔽金属活性中心,防止催化剂失活。此外,较大的空间位阻能够有效地抑制β-H 消除反应,使得产物中高碳烯烃的含量升高。
2.6 溶剂种类对催化乙烯齐聚性能的影响
溶剂对超支化水杨醛亚胺镍系催化剂(Ni-3)催化乙烯齐聚活性和高碳烯烃选择性的影响结果见表2。由表2 可以看出,甲苯为溶剂时,催化活性最高,为1.77×105g/(mol Ni·h)。分析原因为,与甲基环己烷和环己烷相比,甲苯的极性较大,催化剂Ni-3 在甲苯为溶剂的催化体系中溶解性较好,催化活性较高。此外,由表2还可以看出,以甲基环己烷和环己烷为溶剂时,对高碳烯烃的选择性也较低。这可能是由于以甲基环己烷和环己烷为溶剂时会促进链转移反应的发生,乙烯单体倾向于发生二聚反应,导致齐聚产物以丁烯为主。

表1 配体空间位阻对催化乙烯齐聚性能的影响
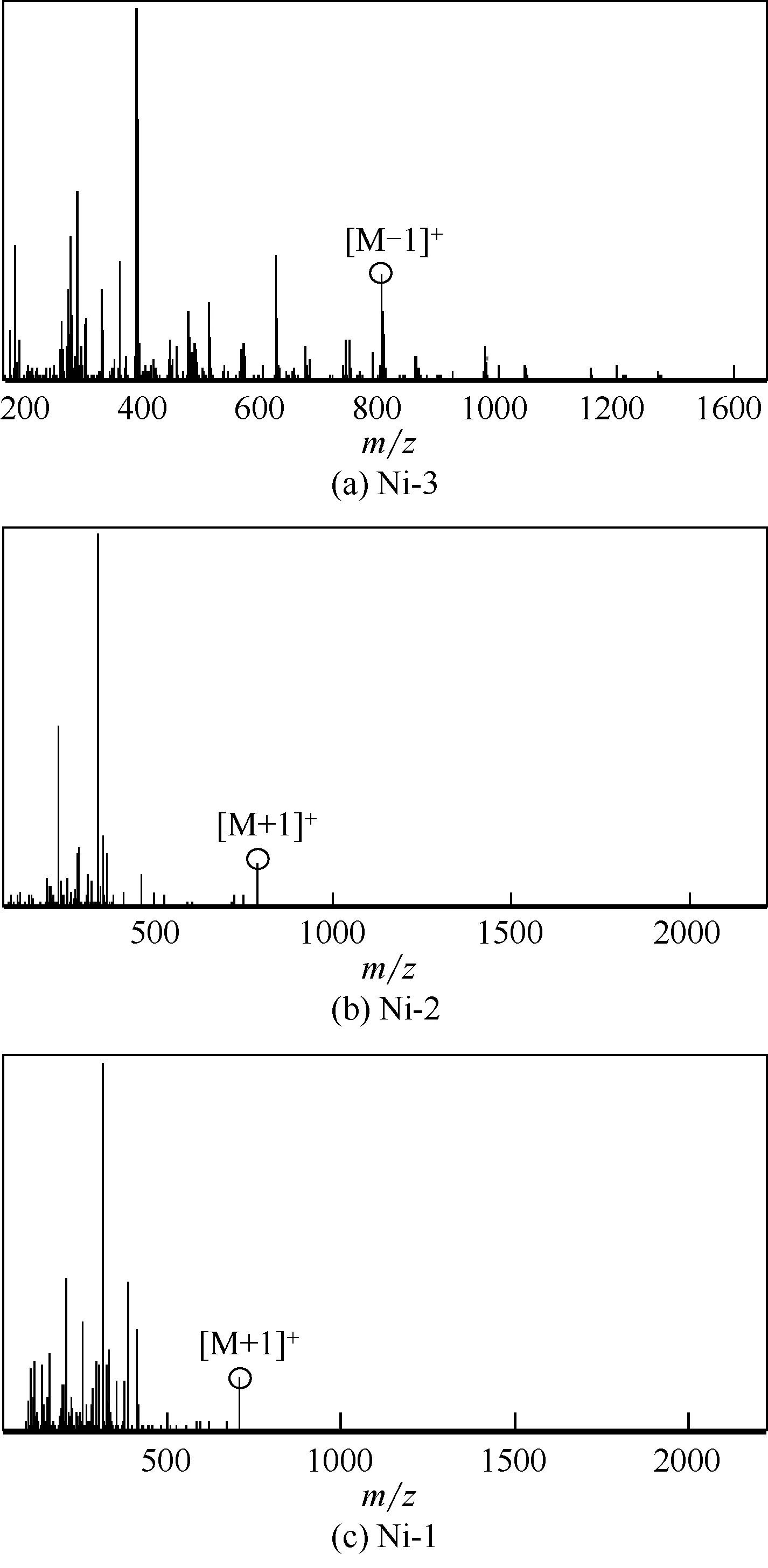
图5 超支化水杨醛亚胺镍系催化剂的质谱
2.7 助催化剂种类对催化乙烯齐聚性能的影响
助催化剂对超支化水杨醛亚胺镍系催化剂(Ni-3)催化乙烯齐聚活性和高碳烯烃选择性的影响结果见表3。由表3可以看出,当采用MAO和Al(Et)3两种不同助催化剂时,超支化水杨醛亚胺镍系催化剂不仅在活性上表现出较大的差异,对产物的选择性也明显不同。当采用Al(Et)3为助催化剂时,催化活性较高,但高碳产物的含量较低。这是因为Al(Et)3较助催化剂MAO 的还原性强,能够将金属镍充分还原成活性中心,催化活性较高。
2.8 反应温度对催化乙烯齐聚性能的影响
反应温度对超支化水杨醛亚胺镍系催化剂(Ni-3)催化乙烯齐聚活性和高碳烯烃选择性的影响结果见表4。由表4 可以看出,催化活性随反应温度增加先升高后降低,25℃时活性最高。原因为反应温度不仅对齐聚速率常数有较大的影响,对乙烯单体在体系中的溶解度和活性中心的稳定性影响也较大,当温度较低时,金属镍不能完全被活化成活性中心,然而较高的温度又会导致催化活性中心分解,催化活性下降[20-21]。同时在较高温度下,乙烯在体系中溶解度下降和活性中心分解速度加快也是导致活性降低的主要原因。此外,随反应温度增加,链转移反应增加速率大于链增长反应增加速率,齐聚产物中高碳烯烃含量下降。
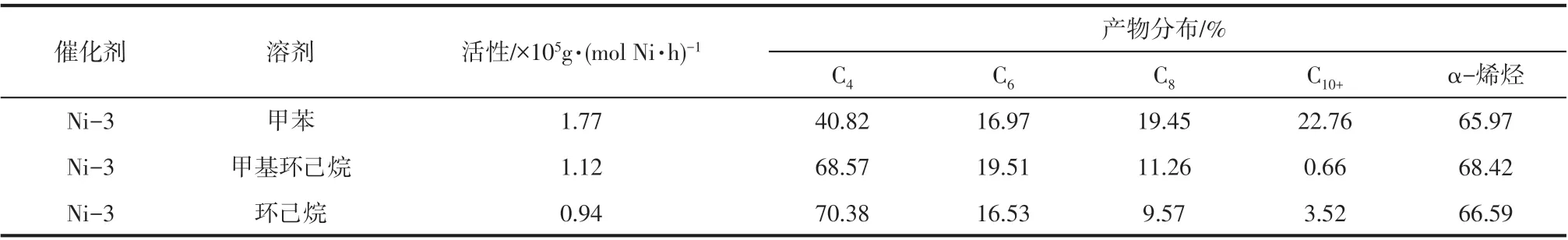
表2 溶剂种类对催化乙烯齐聚性能的影响

表3 助催化剂种类对催化乙烯齐聚性能的影响
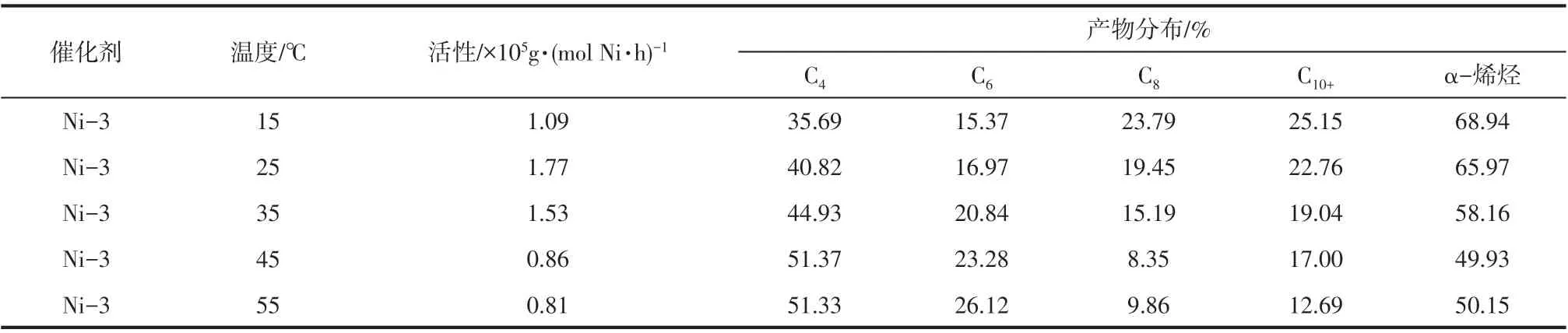
表4 反应温度对催化乙烯齐聚性能的影响
2.9 Al/Ni 物质的量之比对催化乙烯齐聚性能的影响
Al/Ni 物质的量之比对超支化水杨醛亚胺镍系催化剂(Ni-3)催化乙烯齐聚活性和高碳烯烃选择性的影响结果见表5。由表5 可以看出,催化活性随Al/Ni物质的量之比的增大呈现持续升高的趋势。这是因为,助催化剂MAO 能够同时起到消除齐聚体系中水、氧等有害杂质和对金属配合物进行烷基化形成活性中心的双重作用。形成活性中心的数量随Al/Ni 物质的量之比的增加而增大,催化活性升高。此外,从表5还可以看出,催化剂Ni-3对高碳烯烃的选择性随Al/Ni物质的量之比的增加而降低,表明Al/Ni 物质的量之比的升高不利于链增长反应的发生。
2.10 乙烯压力对催化乙烯齐聚性能的影响
乙烯压力对超支化水杨醛亚胺镍系催化剂(Ni-3)催化乙烯齐聚活性和高碳烯烃选择性的影响结果见表6。由表6可以看出,与反应温度和Al/Ni 物质的量之比相比,乙烯压力对催化乙烯齐聚性能的影响较大,催化活性随乙烯压力的增加逐渐升高,当乙烯压力升高到1.0MPa 时,活性可达2.81×105g/(mol Ni·h)。产生这一现象的原因为,乙烯压力增加,气相乙烯分子向甲苯溶剂中扩散的速度增大,乙烯分子在溶剂中的浓度升高,根据反应动力学,乙烯的浓度越大,其与催化剂活性中心配位插入反应的几率越大,催化活性升高[22]。此外,随乙烯压力升高,齐聚产物的碳数分布呈现向高碳烯烃方向移动的趋势,这可能是因为链增长反应速率随乙烯压力增加而增大。
2.11 催化剂结构对催化乙烯齐聚性能的影响
在上述研究工作的基础上,采用Song等[23]合成出的非多支型水杨醛亚胺镍系催化剂和Malgas等[24]合成出的树枝状水杨醛亚胺镍系催化剂(催化剂分子结构如图6)为研究对象,考察催化剂结构对催化乙烯齐聚性能的影响,最佳反应条件下,三种水杨醛亚胺镍系催化剂催化乙烯齐聚结果见表7。由表7可以看出,非多支型镍系催化剂的活性明显高于多支型镍系催化剂,分析原因为,非多支型水杨醛亚胺镍系催化剂在溶剂中具有较好的溶解性。然而,多支型镍系催化剂较大的空间位阻在催化乙烯齐聚过程中能够有效的抑制β-H消除反应的发生,利于高碳烯烃的生成,齐聚产物分子量增大。与超支化水杨醛亚胺镍系催化剂相比,树枝状水杨醛亚胺镍系催化剂对称的分子结构使其具有较大的空间位阻,利于链增长反应的进行,使得高碳烯烃选择性较高,C10+烯烃含量高达100%。
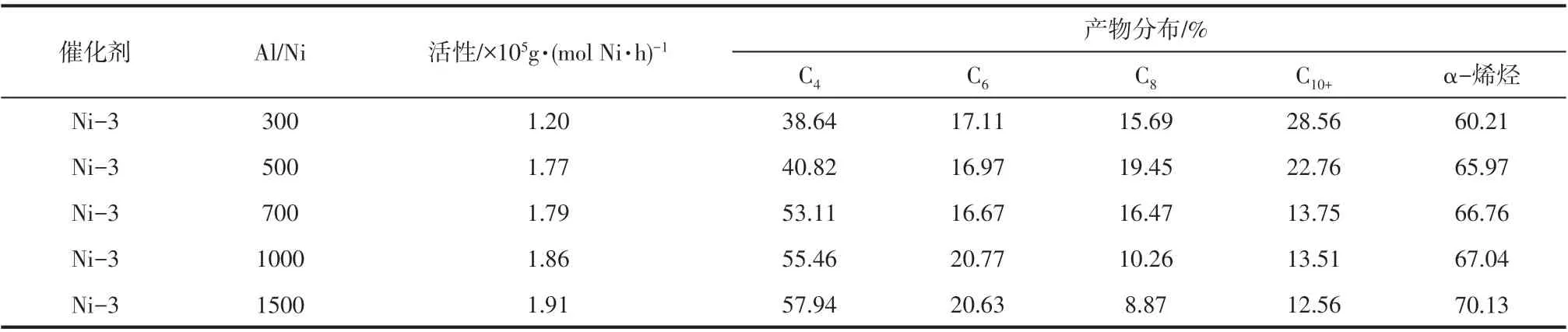
表5 Al/Ni物质的量之比对催化乙烯齐聚性能的影响
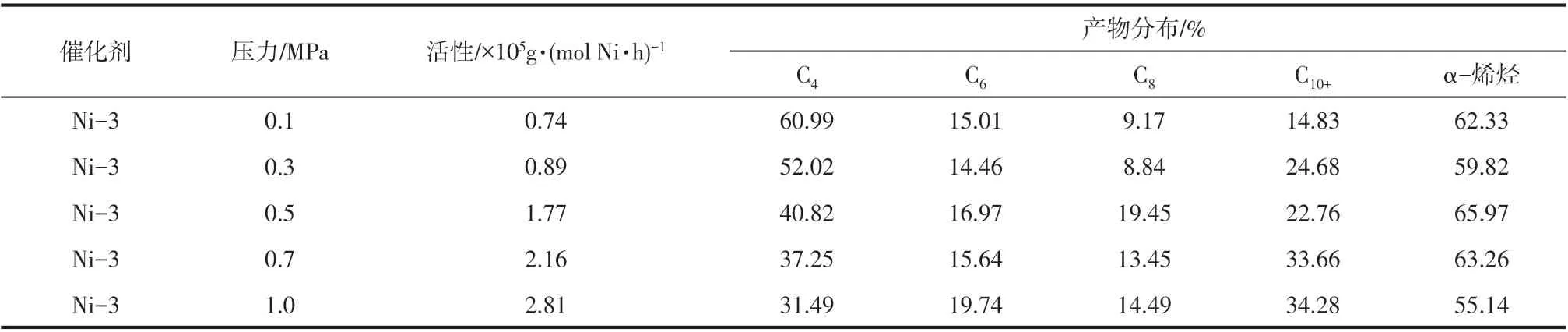
表6 乙烯压力对催化乙烯齐聚性能的影响

表7 催化剂结构对催化乙烯齐聚性能的影响

图6 水杨醛亚胺镍系催化剂的分子结构
2.12 超支化水杨醛亚胺镍系催化剂催化乙烯齐聚机理
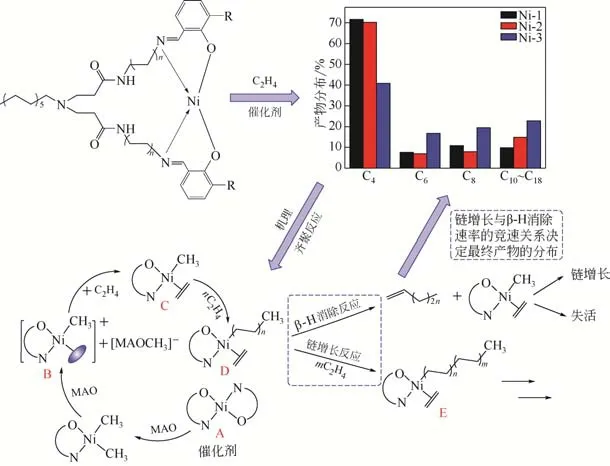
图7 超支化水杨醛亚胺镍系催化剂催化乙烯齐聚机理
根据系列超支化水杨醛亚胺镍系催化剂的化学结构及催化乙烯齐聚产物的分布情况,对其催化乙烯齐聚机理进行推断,如图7所示。系列超支化水杨醛亚胺镍系催化剂催化乙烯齐聚的过程分为四个阶段,分别为催化剂活化、链引发和增长、β-H消除和催化剂失活。首先超支化水杨醛亚胺镍系催化剂在助催化剂MAO 活化下形成具有空轨道的活性中心B;然后反应进入链引发阶段,即乙烯分子与活性中心B发生配位反应,形成烷基金属配合物C;烷基金属配合物C 继续发生链增长反应,形成新的烷基金属配合物D,此时,烷基金属配合物D既可以发生链增长反应,还可以发生β-H 消除反应,当发生链增长反应时,生成具有更长烷基链的烷基金属配合物E,而发生β-H消除反应时,则生成相应碳数的烯烃和烷基金属配合物C。在反应过程中,链增长反应和β-H 消除反应同时进行,两者的竞速关系决定最终产物的分布情况。
3 结论
(1)FTIR、1H NMR、UV-vis、ESI-MS和ICP-MS的分析结果证实合成出的系列具有不同取代基位阻的超支化水杨醛亚胺配体及其镍系催化剂的实际结构与理论设计结构相符。
(2)系列新型超支化水杨醛亚胺镍系催化剂均具有较好的催化乙烯齐聚性能,在MAO 活化下,当反应温度为25℃、乙烯压力为1.0MPa、Al/Ni 物质的量之比为500 时,催化活性可达2.81×105g/(mol Ni·h),C10+烯烃的选择性为34.28%。
(3)超支化水杨醛亚胺配体取代基位阻对其镍系催化剂催化乙烯齐聚活性和产物选择性具有较明显的影响,随着取代基位阻增大,催化活性和C10+烯烃的选择性均呈现升高的变化趋势。
