运动对线粒体稳态调控机制的研究述评
——基于运动介导TFEB调节线粒体质量控制的关键机制探讨
2020-04-09钱帅伟漆正堂丁树哲
钱帅伟,孙 易,漆正堂,丁树哲
(1.武汉体育学院 运动训练监控湖北省重点实验室,湖北 武汉 430079;2.华东师范大学 青少年健康评价与运动干预教育部重点实验室,上海 200241;3.烟台大学 体育学院,山东 烟台 264005)
线粒体具有高度动态可塑性。正常生理情况下,线粒体形态、结构、数量、体积、质量及功能总是维持相对稳定的动态平衡状态,称为线粒体稳态。高度动态稳定的线粒体稳态不仅能保证线粒体高效进行生物氧化和能量转换,进而为细胞生命活动提供绝大部分能量,还能确保线粒体正常参与氧化还原平衡、钙离子稳态调节、细胞信号转导、细胞增殖分化、细胞凋亡和细胞自噬等重要生命过程。但许多内外源应激性刺激(如运动不足、高糖高脂膳食和氧化应激损伤等)均可导致线粒体动态平衡机制失衡(即线粒体稳态调控系统紊乱),致使线粒体健康及细胞健康严重受损,进而可能诱发胰岛素抵抗、代谢综合征、非酒精性脂肪肝、II型糖尿病、癌症、神经退行性疾病和心脑血管疾病等慢性代谢疾病(Gan et al.,2018;Go‐mez-Serrano et al.,2018;Um et al.,2017)。因此,确保线粒体稳态调控系统的功能完整性是维护线粒体健康、细胞健康乃至整体健康的重要基础与前提。
线粒体质量控制是保证线粒体稳态调控系统功能完整性的重要内源性代偿调节机制,主要包括线粒体生物发生、线粒体融合分裂和线粒体自噬等动态协调机制过程(Palikaras et al.,2014)。运动作为一种经典的生理性刺激方式,可诱导线粒体生物发生,增加线粒体数量和体积,产出更多新的健康线粒体,保证运动应激下不同部位对能量的紧急需求(Hood et al.,2016;Little et al.,2011)。运动可高效促进线粒体融合分裂循环的动态平衡,使线粒体形态、结构、长度、大小、数量和分布不断动态更新,进而重构线粒体网络结构(Trewin et al.,2018)。运动还可启动线粒体自噬,加速受损、衰老或非功能线粒体的特异性消化降解,维持线粒体数量、质量及功能完整性(Drake et al.,2019;Hood et al.,2016),并偶联线粒体生物发生、线粒体融合分裂循环等动态机制过程,协同稳定线粒体质量控制,保证线粒体稳态调控系统的功能完整性。但目前普遍认为,关键转录调节因子亦是线粒体质量控制及线粒体稳态调控系统中不可或缺的重要参与者和执行者。运动介导线粒体质量控制的各动态协调机制过程均受到多种关键或重要转录调节因子(如PGC-1α、p53、SIRT1、TFEB等)的严密控制与精细调节,其协同调控机制紊乱可损害线粒体稳态调控系统的功能完整性,进而可能促发慢性代谢疾病(Kim et al.,2017c)。基于此,探讨新型关键转录因子在运动介导线粒体质量控制及线粒体稳态调控中的重要作用,可为运动防控慢性代谢疾病提供新的指导性策略。
转录因子EB(transcription factor EB,TFEB)是近期研究发现的新型关键转录调节因子,是稳定线粒体质量控制,保证线粒体稳态调控系统功能完整性的重要内源性信号调节机制。运动可介导TFEB促进线粒体生物发生,增加线粒体数量和体积,改善线粒体功能,保证线粒体能量供给(Erlich et al.,2018;Mansueto et al.,2017)。运动可能介导TFEB调节线粒体融合分裂循环的动态平衡,进而重构线粒体网状结构,改善线粒体网络的功能完整性(Pastore et al.,2017)。运动还能介导TFEB启动线粒体自噬,靶向降解受损、衰老或非功能线粒体,维持线粒体数量、质量及功能完整性(Erlich et al.,2018;Parousis et al.,2018)。提示,运动通过TFEB介导的线粒体质量控制在维持线粒体稳态调控系统的功能完整性方面具有不可或缺的重要作用。
1 TFEB是溶酶体合成与自噬网络的主控开关
TFEB是碱性螺旋-环-螺旋亮氨酸拉链(bHLH-LZ)转录因子中MITF/TFE家族的重要成员之一。该家族除TFEB外,还包括小眼畸形转录因子(microphthalmia-asso‐ciated transcription factor,MITF)、转录因E3(Transcription factor E3,TFE3)和转录因子 EC(transcription factor EC,TFEC)等转录因子(Kuiper et al.,2004)。TFEB、MITF、TFE3和TFEC均含一个保守bHLH-LZ结构域,可通过该结构域构成同源或异源二聚体,进而发挥转录执行功能(Kuiper et al.,2004)。TFEB主要由476个氨基酸残基构成,包括富谷氨酰胺、螺旋-环-螺旋、亮氨酸拉链和富脯氨酸等模序(Sardiello et al.,2009)。TFEB可通过其转录运行机制,起始相应基因转录,进而调节血管新生、肿瘤发生、溶酶体合成、细胞自噬和线粒体质量控制等诸多病理或生理进程。
TFEB作为近期关注较多的新型转录调节因子,可根据运动、禁食、能量限制、去神经支配、溶酶体功能障碍和肌肉收缩刺激等内外源应激状况,调整其亚细胞定位及转录调节机制,从整体机制上有效控制溶酶体合成和自噬通量水平。基础状态下,TFEB Ser142/Ser211位点被高度磷酸化,其主要以低活性状态存储于胞浆;但内外界应激性刺激可促进TFEB靶向入核,并通过转录控制自噬泡形成与延伸(WIPI、LC3、Atg9)、内容物识别(P62)、自噬溶酶体融合(VPS11、VPS18)和内容物降解(LAMP1、UVRAG)等协同溶酶体表达与调控(coordinated lysosomal expression regulation,CLEAR)组件相关基因表达,促进溶酶体合成和细胞自噬(Settembre et al.,2011b)。一方面,通过上调溶酶体相关基因转录,增加溶酶体数量,增强溶酶体酶活性及功能,促进溶酶体对底物高效降解(Settem‐bre et al.,2011b);另一方面,通过调节自噬不同阶段关键基因表达,从整体机制上正性控制自噬水平(Settembre et al.,2011a,2011b)。TFEB过表达或运动均可促进TFEB靶向入核,改善溶酶体活性及功能,提高自噬体与溶酶体融合率,促进溶酶体内自噬组分高效降解(Mansueto et al.,2017;Settembre et al.,2011a);但RNAi干扰TFEB则可下调自噬通量水平,且其下调程度与不同水平RNAi低聚物所致TFEB干扰程度呈正相关(Settembre et al.,2011a)。
这说明,TFEB可对外源性应激做出快速应答反应,调整其特异性位点的磷酸化状态及功能活性,改变其亚细胞定位及其介导的转录调节机制,在溶酶体合成与自噬调控等方面发挥主控开关作用。基于此,TFEB被普遍认定为溶酶体合成与自噬网络的主控开关(Medina et al.,2015b;Settembre et al.,2011b,2013b)。
2 TFEB关键转录运行机制
TFEB亚细胞定位及转录运行机制受多种磷酸化修饰事件的影响。基础状态下,TFEB主要以磷酸化失活形式聚集在胞浆;当细胞遭受内外源急慢性应激信号刺激时,TFEB去磷酸化激活而靶向入核,激活其下游相关基因转录,发挥其转录执行功能。目前研究已证实,哺乳动物雷帕霉素靶蛋白复合物1(mammalian target of rapamycin complex 1,mTORC1)、蛋白激酶C(protein kinase C,PKC)细胞外信号调节激酶2(extracellular regulated kinase 2,ERK2/MAPK1)均是TFEB上游控制其磷酸化修饰及亚细胞定位的重要信号分子(Ferron et al.,2013;Kim et al.,2017a;Lacombe et al.,2013;Liet al.,2016;Martina et al.,2012,2013;Roczniak-Ferguson et al.,2012;Settembre et al.,2011b,2012;Vega-Rubinde-Celis et al.,2017;Yoneka‐wa et al.,2015)。
2.1 mTORC1/TFEB信号轴
mTORC1是一种在进化上极其保守的丝/苏氨酸蛋白激酶,在细胞生长、分化、增殖、凋亡和自噬等病理或生理进程中具有重要作用。mTORC1可对TFEB进行磷酸化修饰,抑制其胞核定位及其介导的溶酶体合成和细胞自噬。
能量充裕(或氨基酸刺激)时,V-ATPase可与Ragulator相互作用,激活Rag GTPase,使RagA/B与GTP结合,RagC/D与GDP结合,并形成异二聚体,募集mTORC1至溶酶体膜,并被其膜表面Rheb激活,活化的mTORC1可磷酸化TFEB Ser142/Ser211位点,使其与14-3-3蛋白结合,将TFEB阻滞在胞浆并失活,进而抑制溶酶体合成和细胞自噬(Martina et al.,2013;Settembre et al.,2012)。当细胞遭受氧化应激、运动刺激、能量匮乏、溶酶体功能障碍等应激信号刺激时,mTORC1失活,并从溶酶体解离,重新恢复TFEB胞核定位及转录执行功能,促进溶酶体合成和细胞自噬(Martina et al.,2013;Medina et al.,2015b;Roc‐zniak-Ferguson et al.,2012;Settembre et al.,2012)。但近期研究发现,mTORC1不仅能磷酸化TFEB Ser211位点,还可直接磷酸化其Ser122位点,进而抑制其胞核定位及溶酶体合成。而Torin1(mTORC1抑制剂)、氨基酸匮乏、葡萄糖饥饿则可抑制mTORC1及其介导的TFEB Ser122位点磷酸化,促使TFEB靶向入核。但该位点突变时,To‐rin1则不能促进TFEB胞核定位及其介导的溶酶体合成和细胞自噬(Vega-Rubin-de-Celis et al.,2017),提示,TFEB Ser122位点亦是mTORC1控制其亚细胞定位及转录调节机制的重要磷酸化位点。
一些重要分子信号(如 STUB1、MAP4K3、Ca2+、AMPK、FoxO3等)也是控制mTORC1介导TFEB亚细胞定位及转录执行功能的重要因素。STUB1是一种具有E3泛素连接酶活性的胞浆蛋白,在蛋白质量控制中具有重要作用。Sha等(2017)研究发现,基础状态下,mTORC1可磷酸化抑制TFEB,使其以低活性状态存储于胞浆;但应激状态下(如饥饿、mTOR抑制剂等),STUB1可增强其与磷酸化状态TFEB的相互作用,使后者被泛素化,并通过蛋白酶体途径降解,导致非磷酸化TFEB比例增加,进而促进TFEB核定位及其介导的溶酶体合成、自噬体生成和线粒体生物发生。丝裂原蛋白激酶激酶激酶激酶3(mito‐gen-activated protein kinase kinase kinase kinase 3,MAP4-K3)是mTORC1上游氨基酸应激的重要感应分子。Hsu等(2018)研究发现,氨基酸充裕时,MAP4K3可磷酸化HEK 293A细胞TFEB Ser3位点,进而增强Rag GTPases募集TFEB至溶酶体表面的能力,TFEB的溶酶体定位可增强mTORC1对TFEB Ser211位点的磷酸化作用,将其阻滞在胞浆而失活;氨基酸缺乏时,MAP4K3定位溶酶体,并解除其与TFEB的磷酸化作用,重新恢复TFEB核定位及其自噬溶酶体机制。溶酶体内源Ca2+也是调节mTORC1/TFEB信号轴的重要分子信号(Medina et al.,2015a)。营养丰富时,mTORC1可磷酸化抑制TFEB,使其定位于溶酶体膜表面,并阻滞其入核;但饥饿、运动等生理刺激可抑制mTORC1,使溶酶体内源Ca2+经其膜表面的关键Ca2+导通道——粘脂蛋白1(mucolipin 1,MCOLN1)靶向释放至胞浆,进而激活钙调神经磷酸酶(calcineurin,CaN),促进其介导的TFEB去磷酸化及转录激活机制(Medina et al.,2015b)。但药物抑制CaN则可下调运动或饥饿诱导的TFEB转录调节机制(Medina et al.,2015b)。近期,Hood研究团队(Carter et al.,2018a;Kim et al.,2017b;Kim et al.,2019)亦发现,慢性收缩活动可上调骨骼肌TFEB和溶酶体膜标志物LAMP1蛋白表达,并使MCOLN1、Cathepsin D、LAMP2、LAMP2A 等溶酶体蛋白表达同步升高。由于MCOLN1是溶酶体膜表面释放Ca2+的关键通道,推断,肌肉收缩活动/运动很可能促进溶酶体内源Ca2+经MCOLN1靶向释放至胞浆,进而控制mTORC1/TFEB信号轴的活性及功能。腺苷酸活化蛋白激酶(AMP-activated protein kinase,AMPK)是细胞能量状态的敏感感受器。耐力运动可激活AMPK,进而抑制mTORC1,减弱其对TFEB的磷酸化抑制,促进TFEB靶向入核及其介导的溶酶体合成和细胞自噬(Triolo et al.,2019)。叉头框蛋白 O3(forkhead box O3,FoxO3)是近期研究较多的自噬相关转录因子。据报道,FoxO3可增加细胞谷氨酰胺水平,进而抑制mTORC1,促进TFEB胞核定位及介导的溶酶体合成和细胞自噬(Settembre et al.,2013b;van der Vos et al.,2012)。
可知,禁食、能量限制、代谢应激、肌肉收缩活动和运动刺激等急慢性能量应激均可通过直接或间接分子路径调节mTORC1的活性,改变TFEB磷酸化修饰状态、亚细胞定位及其介导的溶酶体合成和细胞自噬(图1)。
2.2 PKC/TFEB信号轴
PKC是哺乳动物中广泛存在的一类磷脂依赖型丝/苏氨酸蛋白激酶家族,在细胞生长、增殖、迁移、分化、凋亡和血管发生等过程中均具有重要作用。根据其结构、功能及作用第二信使的不同,可分为经典型PKC(α,βI,βII,γ)、异常型 PKC(δ,ε,η,θ)和非典型 PKC(ι,ζ,N1-N3)(Sun et al.,2014)。其中,PKCα、PKCβ、PKCδ可通过改变TFEB亚细胞定位及转录调节机制,促进溶酶体合成和细胞自噬。
PKCα、PKCδ主要通过磷酸化修饰糖原合成酶激酶3β(glycogen synthase kinase3β,GSK3β)和 c-Jun 氨基末端激酶(c-Jun N-terminal kinase,JNK)/p38信号通路,进而控制溶酶体合成和自噬通量水平。GSK3β作为细胞中广泛分布的丝/苏氨酸蛋白激酶,在糖原代谢和转录调控中具有重要作用。Li等(2016)研究发现,GSK3β可直接磷酸化TFEB Ser134/Ser138位点,将其阻滞在胞浆而失活,从而抑制溶酶体合成和细胞自噬。但HEP14或其类似物则可通过mTORC1非依赖性途径,直接与PKCα和PKCδ交互作用,使前者转移到胞膜,后者转移到胞膜、胞内囊泡或核膜表面,导致GSK3β磷酸化失活,进而解除对TFEB的磷酸化抑制,致使TFEB靶向入核,从而促进溶酶体合成(Li et al.,2016)。PKCδ还可激发JNK/p38信号的级联激活,增强后者对溶酶体合成和自噬相关基因的抑制因子——ZKSCAN3 Thr153位点的磷酸化,使ZKSCAN3从胞核靶向转位到胞浆而失活,进而解除其对溶酶体合成的抑制作用,协同促进溶酶体合成(Li et al,2016;Saftig et al.,2016)。
PKCβ也是调节TFEB磷酸化修饰及亚细胞定位的重要蛋白激酶。研究发现,PKCβ可直接磷酸化TFEB Ser468、Ser461/Ser462、Ser465/Ser466位点,促进溶酶体合成相关基因表达,进而增加溶酶体数量和体积。但抑制PKCβ则可阻遏TFEB亚细胞定位,下调溶酶体相关基因表达,导致溶酶体功能障碍(Ferron et al.,2013;Lacombe et al.,2013)。
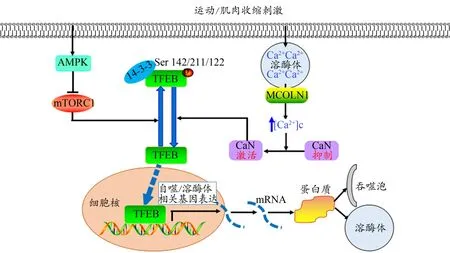
图1 运动/肌肉收缩刺激介导mTORC1控制TFEB亚细胞定位及转录调节机制Figure 1.Subcellular Localization and Transcriptional Regulation Mechanism of TFEB Controlled by Exercise/Muscle Contractile Activity-mediated mTORC1
这说明,PKC(PKCα、PKCβ、PKCδ等)作为细胞中重要的主控调控分子,可通过mTORC1非依赖性途径,控制TFEB亚细胞定位及转录调节机制,促进溶酶体合成和细胞自噬(图2)。
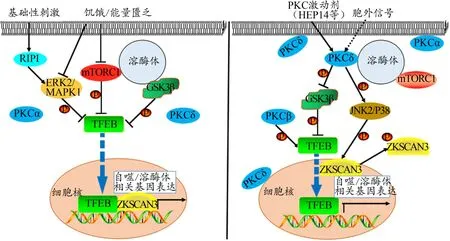
图2 不同刺激介导PKC和ERK2(MAPK1)控制TFEB亚细胞定位及转录调节机制Figure 2.Subcellular Localization and Transcriptional Regulation Mechanism of TFEB Controlled by Stimuli-mediated PKC and ERK2(MAPK1)注:引自Li et al.,2016,略作修改。
2.3 ERK2(MAPK1)/TFEB信号轴
丝裂原活化蛋白激酶(mitogen-activated protein ki‐nase,MAPK)是细胞中一组重要的丝/苏氨酸蛋白激酶,主要包括ERK1/2、JNK、p38 MAPK和ERK5等信号组件。MAPK家族在细胞增殖、分化、转化、凋亡和自噬等病理或生理进程中均具有重要作用。其中,ERK2/MAPK1是控制TFEB亚细胞定位及转录调节机制的关键蛋白激酶(Kaneko et al.,2018;Settembre et al.,2011b)。
基础状态下,ERK2/MAPK1可磷酸化TFEB Ser142位点,使其大量聚集在胞浆;饥饿或能量匮乏时,ERK2/MAPK1可解除其对TFEB的磷酸化抑制,导致TFEB激活及靶向入核,进而转录控制溶酶体合成和自噬相关基因表达(Settembre et al.,2011b)。ERK2/MAPK1上游还存在可对其活性进行正性控制的重要蛋白激酶——受体相互作用蛋白激酶 1(receptor interacting protein kinase 1,RIPK1/RIP1),基础状态下,RIP1可激活ERK2/MAPK1,增强其对TFEB Ser142位点的磷酸化抑制,阻滞TFEB胞核定位及转录调节机制,抑制溶酶体合成和细胞自噬(Yo‐nekawa et al.,2015)。近期研究还发现,AMPK也可直接抑制ERK2/MAPK1的活性,解除后者对TFEB的磷酸化抑制,促使TFEB靶向入核,促进溶酶体合成和细胞自噬(Kim et al.,2017a)。
可知,ERK2/MAPK1是除mTORC1和PKC之外,调节TFEB磷酸化状态及功能活性,进而控制其亚细胞定位及转录调节机制的又一重要蛋白激酶(图2)。
3 运动介导TFEB调节线粒体质量控制及线粒体稳态调控系统的关键内在机制
3.1 运动高效促进TFEB-PGC-1α正反馈共调控回路,诱导线粒体生物发生
线粒体生物发生是细胞内新生线粒体的形成过程。肌肉收缩活动、运动刺激、寒冷、能量限制和禁食等外源性应激均可诱导线粒体生物发生,使线粒体数量增加、体积增大。线粒体生物发生起始于细胞内多起细胞核分子事件,过氧化物酶体增殖物活化受体γ辅激活因子1α(per‐oxisome proliferator-activated receptor-γ coactivator,PGC-1α)被公认为各种急慢性应激诱导线粒体生物发生的万能转录共激活因子。急性运动(Cobley et al.,2012)、急性收缩刺激(Carter et al.,2018b)、耐力运动(Krysciak et al.,2018)、慢性收缩刺激(Parousis et al.,2018)和高强度间歇运动(Little et al.,2011)等诸多运动应激均可诱导PGC-1α表达并靶向转位入核,随后与其下游核呼吸因子1/2(nu‐clear respiratory factor-1/2,NRF1/2)结合,转录激活线粒体转录因子A(mitochondrial transcription factor A,TFAM)和细胞核编码线粒体基因表达,进而促进线粒体生物发生,增加线粒体数量和体积,改善线粒体功能,保障细胞生命活动或运动应激下的能量供给。但运动或肌肉收缩活动通过PGC-1α诱导线粒体生物发生还受多种信号通路在转录后水平的正性控制,如AMP/ATP-AMPK、Ca2+-钙调蛋白依赖性蛋白激酶(calcium/calmodulin-dependent protein kinase,CaMK)、ROS-p38 MAPK-激活转录因子 2(activat‐ing transcription factor 2,ATF2)、NAD+/NADH-沉默信息调节因子1(sirtuin 1,SIRT1)等(Hood et al.,2016)。
TFEB作为溶酶体合成和自噬网络的主控开关,可通过介导PGC-1α表达,诱导线粒体生物发生。Mansueto等(2017)研究发现,C2C12肌细胞TFEB过表达可诱导PGC-1α/β、NRF1、NRF2和TFAM基因表达,促进线粒体生物发生,增加线粒体含量。体内研究亦证实,AAV-TFEB可诱导骨骼肌 PGC-1α/β、PPARα、PPARβ/δ、PPARγ、NRF2 和TFAM等线粒体生物发生或线粒体功能相关基因表达,进而诱导线粒体生物发生(Mansueto et al.,2017)。Erlich等(2018)研究发现,TFEB过表达可促进C2C12肌细胞TFEB及其下游靶基因LC3、LAMP2、P62 mRNA表达,并使线粒体生物发生标志物PGC-1α mRNA表达增加5~6倍,COXIV mRNA表达亦呈升高趋势,提示,TFEB可介导PGC-1α表达,促进线粒体生物发生,增加线粒体数量和体积。Ivankovic等(2016)研究亦发现,SH-SY5Y细胞TFEB过表达可增强PGC-1α mRNA表达,诱导线粒体生物发生,增加线粒体含量,改善线粒体功能。Kim等(2018)研究发现,一氧化碳(CO)致肝细胞线粒体ROS释放可导致蛋白激酶样内质网激酶(protein kinase R-like ER kinase,PERK)激活、Ca2+释放和CaN依赖TFEB胞核转位量增加,并通过介导TFEB-PGC-1α信号轴,协同促进线粒体生物发生,增加线粒体含量。运动也可促进TFEB靶向入核,进而增强PGC-1α表达,诱导线粒体生物发生,增加线粒体数量和体积,改善线粒体功能。Mansueto等(2017)研究发现,急性力竭运动可促进小鼠骨骼肌TFEB靶向入核,但TFEB缺失却可降低运动耐力水平,特异性激活骨骼肌TFEB则可重新改善其运动耐力水平。该运动应答机制很可能与TFEB介导的线粒体生物发生水平上调有关。有研究发现,运动、禁食均可促进骨骼肌、肝脏TFEB靶向入核,进而调控脂代谢、脂肪酸氧化和生酮相关基因表达,其与TFEB介导PGC-1α表达密切相关;但TFEB缺失则可减弱饥饿诱导的PGC-1α表达(Medina et al.,2015b;Settembre et al.,2013a)。提示,PGC-1α很可能部分受TFEB控制。这说明,运动可通过TFEB介导PGC-1α表达,促进线粒体生物发生,增加线粒体含量,改善线粒体功能。
但也有研究表明,PGC-1α亦可作为TFEB上游重要的分子调节信号。过表达TFEB或PGC-1α均可显著改善小鼠脑神经瘤细胞线粒体功能,抑制氧化应激损伤,减少突变亨廷顿蛋白(mutant huntingtin,mHtt)聚集;但沉默细胞TFEB的同时,过表达PGC-1α,则不能减少mHtt等毒性蛋白清除(Tsunemi et al.,2012)。提示,PGC-1α可作为TFEB上游的关键信号,并通过转录共激活TFEB,增强线粒体功能和自噬溶酶体功能,降解胞浆毒性蛋白。Man‐sueto等(2017)研究发现,PGC-1α-/-小鼠骨骼肌TFEB表达较低且主要分布于胞浆,但AAV-TFEB过表达可促进PGC-1α-/-小鼠骨骼肌 NRF1、NRF2 和 TFAM 基因表达,增加线粒体体积和密度。急性运动亦可促进PGC-1α-/-小鼠骨骼肌TFEB表达及靶向入核,进而增强NRF1/2、PPARα/β/δ等线粒体生物发生相关基因表达,提高运动耐力水平(Mansueto et al.,2017)。提示,PGC-1α可作为TFEB上游重要的激动信号,或运动可介导TFEB通过PGC-1α非依赖性途径,促进线粒体生物发生。Erlich等(2018)研究发现,基础状态下,PGC-1α-/-小鼠骨骼肌TFEB和TFE3蛋白含量较野生型小鼠分别下降30%和50%,急性运动虽可使野生型小鼠骨骼肌TFEB转录水平增加2~3倍,核转位量增加4倍,但该运动应答机制在PGC-1α-/-小鼠骨骼肌中并未得到显著响应。提示,PGC-1α缺失可能是PGC-1α-/-小鼠骨骼肌TFEB表达下调的主要原因。
基于TFEB和PGC-1α在转录共调控方面的同向交互作用特性,可知,二者在运动介导线粒体生物发生等方面具有动态协调性,甚至形成正反馈共调控回路,协同促进线粒体生物发生。Vainshtein等(2015a)研究认为,敲除PGC-1α可降低小鼠骨骼肌TFEB蛋白含量及核转位量,降低线粒体生物发生和线粒体自噬,导致肌肉萎缩及肌肉质量下降;但过表达PGC-1α则可增加骨骼肌TFEB蛋白含量,促进Cathepsin D、LAMP2蛋白表达,增加线粒体含量,改善线粒体自噬功能。该研究还认为,TFEB与PGC-1α在调节线粒体生物发生、溶酶体合成和线粒体自噬等方面具有动态协同性,甚至形成正反馈共调控回路(Vainshtein et al.,2015a)。Scott等(2014)研究亦认为,TFEB与PGC-1α可构建正反馈共调控回路,共同维持线粒体生物发生与线粒体自噬的动态循环平衡,且很可能受GCN5L1负性控制。Parousis等(2018)研究发现,慢性收缩活动可同步增强C2C12肌细胞TFEB和PGC-1α表达,协同促进溶酶体合成和线粒体生物发生,改善线粒体功能和自噬溶酶体功能。这说明,运动或收缩活动可作为外源性应激因素,正性控制TFEB-PGC-1α正反馈共调控回路的动态循环平衡,协同促进线粒体生物发生、溶酶体合成和线粒体自噬。
以上研究表明,TFEB与PGC-1α在转录共调控方面具有交互性、协同性、动力性和系统性,甚至构成正反馈共调控回路。运动可高效促进TFEB-PGC-1α共调控回路的动态循环平衡,进而诱导线粒体生物发生,增加线粒体数量和体积,改善线粒体功能,快速高效应答运动应激下不同部位对能量的紧迫需求。
3.2 运动可能介导TFEB调节线粒体融合分裂循环的动态平衡
线粒体融合分裂循环是继线粒体生物发生后的又一起线粒体事件,是决定线粒体形态可塑性及网状结构的关键因素。线粒体通过融合分裂的动态循环进行组分重构,使其形态、结构、数量、长度、分布和质量不断动态调整更新,将受损、衰老或非功能线粒体特异性隔离出线粒体网络,并进行靶向修复或清除,进而维持线粒体网络的功能完整性,快速应答运动应激致能量代谢环境急剧变化时不同部位对能量的紧急需求。线粒体融合事件的关键执行蛋白主要有线粒体融合蛋白1(mitofusin-1,MFN1)、MFN2和视神经萎缩症蛋白1(optical atrophy-1,OPA1)等(Malka et al,2005)。MFN1、MFN2主要介导线粒体外膜融合,OPA1处于内外膜之间,主要负责线粒体内膜融合(Malka et al.,2005)。MFN1、MFN2在促进线粒体融合方面既相互联系,又相互区别。MFN1功能较单一,其GTPase活性较MFN2高,能更高效融合线粒体;MFN2则是一个多功能分子,不仅能高效融合线粒体,在线粒体代谢和细胞凋亡等进程中亦发挥重要作用(Chen et al.,2003;Ishihara et al.,2004)。线粒体分裂事件的关键执行蛋白主要有动力相关蛋白1(dynamin-related pro‐tein 1,DRP1)、线粒体分裂蛋白 1(mitochondrial fission protein-1,FIS1)和线粒体分裂因子(mitochondrial fission factor,MFF)等(Malka et al.,2005)。正常情况下,DRP1主要以可溶性形式散布于胞浆;线粒体分裂时,均匀定位于线粒体外膜的FIS1、MFF可将胞浆可溶性蛋白DRP1招募至线粒体外膜,协同促进线粒体分裂,导致线粒体碎裂化和片段化(Kim et al.,2007)。运动所致能量代谢环境的急剧变化可高效促进线粒体融合分裂循环的动态调整。目前普遍认为,耐力训练可增强线粒体融合蛋白MFN2、OPA1表达,降低DRP1 Ser616磷酸化水平,进而促进线粒体融合,抑制线粒体分裂(Arribat et al.,2019;Axelrod et al.,2018;Fealy et al.,2014;Iqbal et al.,2013);去神经支配、急性运动可抑制线粒体融合蛋白MFN2、OPA1表达,促进DRP1 Ser616磷酸化水平,进而抑制线粒体融合,促进线粒体分裂(Iqbal et al.,2013;Kruse et al.,2017;Moore et al.,2018;Pagano et al.,2014)。但亦有研究认为,线粒体分裂蛋白DRP1缺失可降低肌肉长时运动耐力及运动适应能力(Moore et al.,2019);急性运动也可促进线粒体融合蛋白MFN2表达,进而改善线粒体功能(Busquets-Cortes et al,2017)。提示,运动可使线粒体融合与分裂蛋白表达均同步上调,进而引起更高水平的线粒体融合分裂循环的动态平衡。
目前有关TFEB与线粒体融合分裂循环关键蛋白交互作用的研究较少(Erlich et al.,2018)。Demers-Lamarche等(2016)研究发现,成纤维细胞敲除OPA1可诱发线粒体功能障碍和ROS大量产生,进而破坏溶酶体酸性环境,抑制TFEB转录控制的溶酶体酸性磷酸酶(lysosomal acid phosphatase,LAP)、溶酶体酸脂酶(lysosomal acid lipase,LAL)和Cathepsin B的活性,导致溶酶体功能障碍,致使非功能线粒体等未降解底物聚集。提示,线粒体内膜融合蛋白OPA1缺失可诱发线粒体功能障碍,进而抑制TFEB介导的溶酶体合成。Santin等(2016)研究发现,单胺氧化酶A(MAO-A)过表达可抑制TFEB胞核定位,并使Cathepsin D、LAMP1等溶酶体蛋白聚集,进而损害溶酶体功能;MAO-A还可使线粒体DRP1、Parkin移位,导致线粒体分裂,并使LC3-II、P62和泛素化蛋白聚集,自噬进程受阻,进而诱发心肌细胞死亡。但过表达TFEB则可减少自噬体聚集,阻止线粒体分裂,抑制心肌细胞死亡和心力衰竭(Santin et al.,2016)。提示,TFEB在抑制DRP1介导的线粒体分裂过程中具有重要作用。Chen等(2018)研究发现,心肌LRP6缺失可导致DRP1/mTORC1信号激活及其下游TFEB核定位障碍,进而抑制自噬降解和脂肪酸利用;而DRP1抑制剂Mdivi-1则可抑制mTORC1,促进TFEB靶向入核,进而增强LC3B介导的自噬降解及PPARα、PPARδ介导的脂肪酸利用,改善心力衰竭或心功能。提示,DRP1亦可作为TFEB上游的负性调节信号,抑制TFEB及其介导的溶酶体合成和细胞自噬。但TFEB可否控制OPA1介导的线粒体内膜融合,TFEB与MFN1/2、FIS1和MFF等其他线粒体融裂蛋白的交互作用却未明晰,有待进一步研究。
目前,运动介导TFEB对线粒体融合分裂循环的调控作用及机制知之甚少。Pastore等(2017)研究发现,作为与TFEB同家族同功能的另一转录分子,TFE3与TFEB可协同控制饮食或运动应激下的葡萄糖代谢、脂肪酸氧化、线粒体动力学和线粒体功能等诸多代谢过程。急性力竭运动可促进野生型小鼠肝脏TFEB、TFE3靶向入核,使其核蛋白含量同步增加,而TFE3的表达又可增强肝脏FIS1、DRP1、OPA1、MFN1和MFN2基因表达,进而高效促进线粒体融合分裂循环,改善线粒体功能;但敲除肝脏TFE3则可显著下调肝脏FIS1、DRP1和MFN1基因表达,抑制线粒体融合分裂能力,降低急性力竭运动的耐力水平(Pastore et al.,2017)。提示,运动可介导TFE3激发更高水平的线粒体融合分裂循环的动态平衡,进而重构线粒体网状结构。但遗憾的是,该研究并未明晰TFEB在运动介导线粒体融合分裂循环中的作用。近期有研究发现,6周耐力跑台运动可激活高脂小鼠骨骼肌TFEB及其转录调节机制,增强线粒体融合蛋白MFN1表达,但对MFN2、DRP1等其他线粒体蛋白表达均无显著影响(钱帅伟,2018)。该研究表明,运动介导TFEB仅影响线粒体融合分裂的部分蛋白表达,故其对线粒体融合分裂循环的动态平衡可能未产生整体性影响。
以上研究表明,运动虽可介导TFEB影响线粒体融合分裂循环的动态平衡,进而调整线粒体网状结构,但可能未对其产生整体性影响。但有关TFEB与线粒体融合分裂循环分子事件的研究仍较少,其具体调控机制还需进一步探讨。
3.3 运动介导TFEB启动线粒体自噬,稳定线粒体质量控制及线粒体稳态调控系统
线粒体自噬是继线粒体生物发生、线粒体融合分裂循环后,维护线粒体质量控制及线粒体稳态的又一重要代谢装置。线粒体动力学变化所致亲代线粒体的动态变化分裂最终产生2个不均匀的子代线粒体。其中,膜电位严重下降的子代,以及超过自身修复能力的子代,可通过线粒体自噬途径特异性降解。研究已证实,急性运动(Vain‐shtein et al.,2015b)、急性收缩活动(Erlich et al.,2018)、耐力训练(Chen et al.,2018)、慢性收缩活动(Parousis et al.,2018)、能量限制(Zhao et al.,2018)和去神经支配(Vainshtein et al.,2015a)等内外源急慢性应激均可启动线粒体自噬,选择性降解去极化损伤线粒体,稳定线粒体质量控制,确保线粒体稳态调控系统的功能完整性。但运动介导线粒体自噬维护线粒体质量控制及线粒体稳态的具体调控机制并未完全明晰。TFEB作为溶酶体合成和自噬网络的主控开关,可在运动应激下激发线粒体自噬,进而在线粒体质量控制及线粒体稳态调控中发挥重要作用。
3.3.1 运动介导TFEB启动线粒体自噬的PINK1/Parkin信号机制
PINK1/Parkin介导的线粒体自噬在破损或衰老线粒体降解中具有重要作用(Clark et al.,2006)。PINK1是一种靶向线粒体的丝/苏氨酸蛋白激酶,是线粒体膜极化状态的敏感传感器。基础状态下,PINK1由胞核基因编码并在胞质合成后,通过线粒体膜间隙,进入线粒体内部,被线粒体蛋白水解酶降解(Matsuda et al.,2010)。但线粒体损伤或线粒体应激事件可使线粒体内膜电位消散,PINK1从外膜转运至内膜的跨膜机制障碍,不能被线粒体蛋白水解酶降解,最终稳定并大量聚集在线粒体外膜(Matsu‐da et al.,2010)。外膜累聚的PINK1可通过Parkin非依赖性途径,直接募集自噬体,启动微弱的线粒体自噬(Laz‐arou et al.,2015);也可募集并磷酸化激活 Parkin(Ser 65位点),使其选择性转位至受损线粒体,而后介导MFN1/2、VDAC1和DRP1等蛋白泛素化,使自噬体包裹线粒体,启动线粒体自噬,降解受损严重的线粒体(Fivenson et al.,2017)。同时,自噬接头蛋白P62亦被募集到线粒体,并与LC3-II结合,参与启动线粒体自噬(Geisler et al.,2010)。PINK1/Parkin下游还有一个重要的泛素化底物PARIS。Parkin可与PARIS结合,介导后者泛素化降解,其功能缺失可致PARIS蛋白大量聚集,损伤线粒体降解机制障碍(Shin et al.,2011)。PARIS亦具有转录因子活性,可与PGC-1α基因启动子DNA序列结合而抑制该基因表达。PARIS累积所致下游靶基因PGC-1α表达下调可能是Par‐kin功能缺失致帕金森症的重要因素之一(Shin et al.,2011)。
目前关于TFEB与PINK1/Parkin信号交互作用,进而调节线粒体自噬的研究还较少。Kim等(2018)研究发现,CO致肝细胞线粒体ROS释放可通过PERK-Ca2+-CaNTFEB-PGC-1α信号轴,协同促进溶酶体合成、线粒体生物发生和PINK1/Parkin介导的线粒体自噬,进而改善线粒体功能;但采用siRNA沉默TFEB时,CO虽不能影响线粒体PINK1蛋白表达,却可极显著减少线粒体定位的Parkin蛋白含量。提示,TFEB可作为Parkin(而非PINK1)上游重要的激活信号,募集Parkin至线粒体片段,启动线粒体自噬。但更多研究认为,PINK1/Parkin可作为TFEB上游的关键信号机制,使后者转位入核,增强溶酶体合成能力及自噬溶酶体功能,进而对线粒体自噬进行特异性控制。例如,Ivankovic等(2016)研究发现,线粒体解偶联剂CCCP可激活神经母细胞瘤SH-SY5Y细胞PINK1/Parkin信号机制,进而促进P62 mRNA和蛋白表达,诱导线粒体自噬;沉默PINK1则可抑制P62蛋白表达、溶酶体蛋白表达及线粒体自噬活性。CCCP亦可介导SH-SY5Y细胞PINK1/Parkin通路,促进TFEB靶向入核,增强其靶基因P62 mRNA表达,增加葡萄糖脑苷脂酶(glucocerebrosi‐dase,GCase)和Cathepsin D活性,促进其共激活因子PGC-1α mRNA表达,进而改善溶酶体活性及功能,增加线粒体含量,增强线粒体自噬能力,维持线粒体质量控制及线粒体稳态(Ivankovic et al.,2016)。Nezich等(2015)研究发现,PINK1/Parkin介导的线粒体自噬途径中,由于对自噬蛋白和溶酶体蛋白需求量急剧增加,而这些蛋白的基因表达取决于MITF/TFE家族(如TFEB、TFE3和MITF等)活性,TFEB能以PINK1/Parkin依赖性方式转位至胞核,但其活化及胞核转位一般需要Parkin、Atg5和Atg9A的交互作用,并可反式激活多种自噬溶酶体基因表达,进而降解损伤或衰老线粒体。Zhang等(2017)研究亦发现,PINK1/Parkin机制可促进神经细胞TFEB靶向入核,进而转录激活自噬溶酶体基因表达,特异性降解损伤线粒体,改善线粒体功能,防治神经退行性病变。Siddiqui等(2015)研究发现,Parkin Q311X突变所致Parkin功能缺失可使其泛素化底物PARIS降解机制障碍,PARIS蛋白大量聚集,通过抑制PGC-1α-TFEB核转位信号,导致溶酶体合成能力及溶酶体功能降低,线粒体功能及损伤线粒体降解机制障碍,线粒体质量控制及线粒体稳态功能紊乱,导致神经退行性病变。但雷帕霉素(rapamycin)可通过Parkin非依赖性途径直接激活TFEB,重新恢复PGC-1α-TFEB核转位信号,稳定线粒体质量控制及线粒体稳态调控的功能完整性,改善神经退行性病变体征(Siddiqui et al.,2015)。提示,PINK1/Parkin或Parkin/PARIS可作为PGC-1α-TFEB正反馈共调控回路上游重要的分子信号,调节自噬溶酶体机制、线粒体质量控制及线粒体稳态调控系统。
运动或收缩活动是促进PINK1/Parkin介导线粒体自噬的经典性生理刺激模式,但其具体分子调节机制并未完全阐明。Parousis等(2018)研究发现,慢性收缩活动可协同增强C2C12肌细胞 PINK1、PGC-1α、TFEB和Cathep‐sin D等蛋白表达,并使PINK1、PGC-1α、P62 mRNA表达增加1.5~2倍,进而促进溶酶体合成、线粒体生物发生和线粒体自噬,改善线粒体功能和自噬溶酶体功能。提示,收缩活动很可能激活PINK1/Parkin通路,促进PGC-1α-TFEB正反馈共调控回路的动态循环平衡,改善线粒体自噬功能,进而稳定线粒体质量控制和线粒体稳态调控系统。线粒体外膜蛋白VDAC1与自噬接头蛋白P62均是PINK1/Parkin介导线粒体自噬途径中必不可少的重要信号分子(Geisler et al.,2010)。Erlich等(2018)研究发现,急性收缩刺激5 h可促进C2C12肌细胞VDAC1与P62共定位,进而诱导线粒体自噬,其很可能与PGC-1α驱动TFEB靶向入核有关。Hood研究团队(Chen et al.,2018;Vainshtein et al.,2015b)发现,急性力竭运动可显著增强骨骼肌线粒体自噬标志物LC3-II、PINK1、Parkin和P62蛋白表达,抑制PARIS蛋白表达,促进PINK1/Parkin/PARIS信号轴介导的线粒体自噬,并认为其很可能与PGC-1α驱动TFEB靶向入核密切相关(Erlich et al.,2018;Vainshtein et al.,2015b)。这说明,运动极有可能激活PINK1/Parkin机制,进而抑制PARIS,解除其对PGC-1α的抑制效应,增强PGC-1α-TFEB核转位信号,形成正反馈共调控回路,特异性高速降解损伤或衰老线粒体,确保线粒体质量控制及线粒体稳态调控系统的稳定性和完整性。
可知,运动介导PINK1/Parkin通路调节线粒体质量控制及线粒体稳态调控系统的线粒体自噬机制至少体现在以下3个方面:1)运动介导PINK1/Parkin通路通过TFEB非依赖的传统经典信号机制,诱导线粒体自噬;2)运动介导PINK1/Parkin机制促进TFEB靶向入核,或与PGC-1α构建PGC-1α-TFEB正反馈共调控回路,协同促进线粒体自噬;3)运动介导PINK1/Parkin机制抑制PARIS,解除其对PGC-1α的抑制作用,激活PGC-1α-TFEB核转位信号,形成正反馈共调控回路,协同启动线粒体自噬。
3.3.2 运动介导TFEB启动线粒体自噬NIX/BNIP3信号机制
NIX/BNIP3机制是运动介导线粒体自噬的另一条经典信号调节通路。NIX/BNIP3主要位于线粒体外膜,可直接与自噬蛋白LC3-II相互作用,募集自噬体包裹并降解损伤或衰老线粒体,以PINK1/Parkin非依赖途径启动线粒体自噬(Hamacher-Brady et al.,2016)。NIX也可与Parkin交互作用,招募DRP1与Parkin定位去极化损伤的线粒体,促进线粒体分裂,触发线粒体自噬(Ding et al.,2010)。近期研究发现,NIX同源物BNIP3可与PINK1相互作用,抑制后者裂解,使其累聚在线粒体外膜,并募集Parkin,诱导线粒体自噬(Zhang et al.,2016)。这说明,NIX/BNIP3可通过与LC3-II蛋白直接作用,或通过与PINK1/Parkin协同作用等分子路径,促进线粒体自噬。
但目前关于TFEB与NIX/BNIP3上下游调控关系及交互作用机制,以及二者在线粒体自噬调控中的作用及分子机制的研究较少。Ma等(2012)研究发现,心肌BNIP3过表达可使自噬体过度聚集,溶酶体功能耗竭,致使心肌细胞死亡;但过表达TFEB可增加溶酶体含量和自噬溶酶体数量,阻止P62蛋白聚集,降解去极化损伤线粒体,抑制BNIP3介导的心肌细胞凋亡。Ma等(2015)进一步发现,心肌缺血再灌注可导致ROS产生和BNIP3表达上调,使Beclin1过度聚集,mTORC1被激活,TFEB核转位机制抑制,溶酶体功能降低,去极化损伤线粒体的自噬降解机制障碍,致使心肌细胞死亡。局部敲低Beclin1可抑制mTORC1,激活TFEB及其介导的PGC-1α表达,促进溶酶体合成、线粒体生物发生和线粒体自噬,清除去极化损伤线粒体,维持线粒体质量控制,抑制BNIP3及缺氧复氧所致心肌细胞死亡;但过表达Beclin1则可激活mTORC1、抑制TFEB核转位机制,导致溶酶体数量减少及PGC-1α转录机制抑制(Ma et al.,2015)。提示:1)尽管BNIP3在介导细胞凋亡的同时,也可启动线粒体自噬,并作为一种适应性保护机制,清除受损或去极化线粒体,但由于其功能活性或信号较弱,并不能逆转BNIP3介导的细胞凋亡;2)促凋亡蛋白BNIP3可能抑制PGC-1α-TFEB核转位信号,降低溶酶体合成、线粒体生物发生和自噬溶酶体功能,但激活TFEB却可恢复PGC-1α-TFEB核转位信号,促进溶酶体合成和线粒体生物发生,增强自噬溶酶体功能,特异性降解去极化损伤线粒体,抑制BNIP3及其介导的细胞凋亡。
尽管上述研究从细胞凋亡视角对TFEB与NIX/BNIP3的上下游调控关系及交互作用进行了机制性探释,但近期,以癌症恶病质、运动和收缩活动为实验模型,以细胞自噬和线粒体自噬为主要研究视角的实验证据似乎并不支持TFEB与NIX/BNIP3之间存在交互抑制作用的研究论断。例如,Aversa等(2016)研究发现,癌症恶病质患者骨骼肌Beclin1、LC3B-II、P62和Parkin等自噬蛋白表达均显著较高,但线粒体自噬蛋白BNIP3和NIX/BNIP3L,以及TFEB核蛋白含量仅呈微弱增高的同步变化趋势,研究认为,溶酶体降解功能下降可能是自噬体降解机制障碍、线粒体自噬功能受损的重要原因。Smiles等(2016)研究发现,同期进行抗阻和耐力运动后摄入酒精可诱导骨骼肌细胞凋亡,降低肌肉蛋白质合成;但蛋白质摄入则可增加骨骼肌p53、TFEB和PGC-1α等转录因子核蛋白含量,并上调线粒体生物发生标志物TFAM、SCO2和NRF1 mRNA表达,以及COX-IV、ATPAF1和VDAC1蛋白表达,促进BNIP3 mRNA和蛋白表达,进而诱导线粒体生物发生和线粒体自噬,改善线粒体质量控制,维持骨骼肌代谢功能稳态。Kwon等(2018)研究发现,8周抗阻训练在激活骨骼肌Akt/mTOR/p70S6K信号通路,促进肌肉蛋白质合成,使骨骼肌肥大的同时,还可下调骨骼肌p-AMPK Thr172、LC3-II和Cathepsin L蛋白表达,增加P62蛋白表达,但并不影响TFEB、LAMP2和BNIP3蛋白表达,致使细胞自噬和线粒体自噬保持基础活性,进而抑制肌肉蛋白质降解,维持骨骼肌质量及功能。Jang等(2018)研究发现,6周有氧耐力训练可增强PD小鼠大脑黑质LC3-II、P62、Beclin1、BNIP3、LAMP2、TFEB和Cathepsin L等自噬溶酶体蛋白表达,促进溶酶体合成、细胞自噬和线粒体自噬,稳定线粒体质量控制及线粒体稳态,改善PD相关病理症状。Cart‐er等(2018a)研究发现,衰老可显著上调骨骼肌TFEB及其下游溶酶体标志物LAMP2、Cathepsin D等蛋白表达,并使LAMP1蛋白表达呈微弱升高趋势,NIX/BNIP3介导的线粒体自噬活性呈显著增强态势;而慢性收缩活动/慢性运动却可抑制衰老骨骼肌TFEB介导的溶酶体合成及自噬溶酶体机制,降低线粒体自噬能力,改善衰老骨骼肌质量及器官整体功能。可知,尽管以癌症恶病质、运动和收缩活动为研究模型,以细胞自噬和线粒体自噬为主要研究视角的多项实验研究仍未明晰TFEB与NIX/BNIP3的具体上下游调控关系及交互作用机制,但至少已证实,TFEB与NIX/BNIP3通过运动介导的线粒体自噬对线粒体质量控制及线粒体稳态的调节具有同步变化效应或协同作用特性,并不存在交互抑制效用。
这说明,TFEB与NIX/BNIP3通过运动介导的线粒体自噬对线粒体质量控制及线粒体稳态调控系统的调节具有同步变化效应或协同作用特性。但TFEB与NIX/BNIP3具体上下游分子调控关系,及其在线粒体自噬调控、线粒体质量控制及线粒体稳态调控系统中的作用仍不明确,有待进一步论证。
4 小结与展望
线粒体稳态调控系统的功能完整性离不开线粒体质量控制,即线粒体生物发生、线粒体融合分裂循环和线粒体自噬等动态协调机制过程。运动可介导TFEB的去磷酸化激活及靶向入核,并与受AMP/ATP/AMPK、Ca2+/CaMK、ROS/p38 MAPK、NAD+/NADH/SIRT1等信号通路正性控制的万能转录共激活因子——PGC-1α协同作用,构建PGC-1α-TFEB正反馈共调控回路,随后辅激活NRF1/2等核转录因子,进而激活一系列核基因编码的线粒体蛋白基因序列,诱导线粒体生物发生,增加线粒体数量和体积,快速应答运动应激下不同部位对能量的紧急需求,且该机制在整个线粒体事件中可能占据极其重要的地位。线粒体融合分裂循环是继线粒体生物发生后的又一起线粒体事件,是决定线粒体形态可塑性及网状结构的重要因素。运动可介导TFEB调节线粒体融合分裂循环的动态平衡,进而影响线粒体网络的功能完整性,但目前研究尚未支持其对线粒体融合分裂循环的整体性影响。该分子事件尚知之甚少,具体调控机制需进一步探讨。线粒体自噬是继线粒体生物发生、线粒体融合分裂循环后,维护线粒体质量控制的又一重要内源性代谢装置。就PINK1/Parkin机制而言,运动不仅能介导PINK1/Parkin机制促进TFEB靶向入核,并构建PGC-1α-TFEB正反馈共调控回路,促进线粒体自噬;还能激活PINK1/Par‐kin/PARIS/PGC-1α信号通路,形成PGC-1α-TFEB共调控回路,诱导线粒体自噬。就NIX/BNIP3机制而言,运动可介导NIX/BNIP3与TFEB同步或协同作用,促进线粒体自噬。经上述信号通路启动的线粒体自噬,可特异性高效降解受损、衰老或非功能线粒体,进而维护线粒体质量控制及线粒体稳态。这说明,运动可介导TFEB构建线粒体生物发生、线粒体融合分裂循环和线粒体自噬等动态协调过程的信号联动机制,进而稳定线粒体质量控制,确保线粒体稳态调控系统的功能完整性,防控慢性代谢疾病发生(图3)。
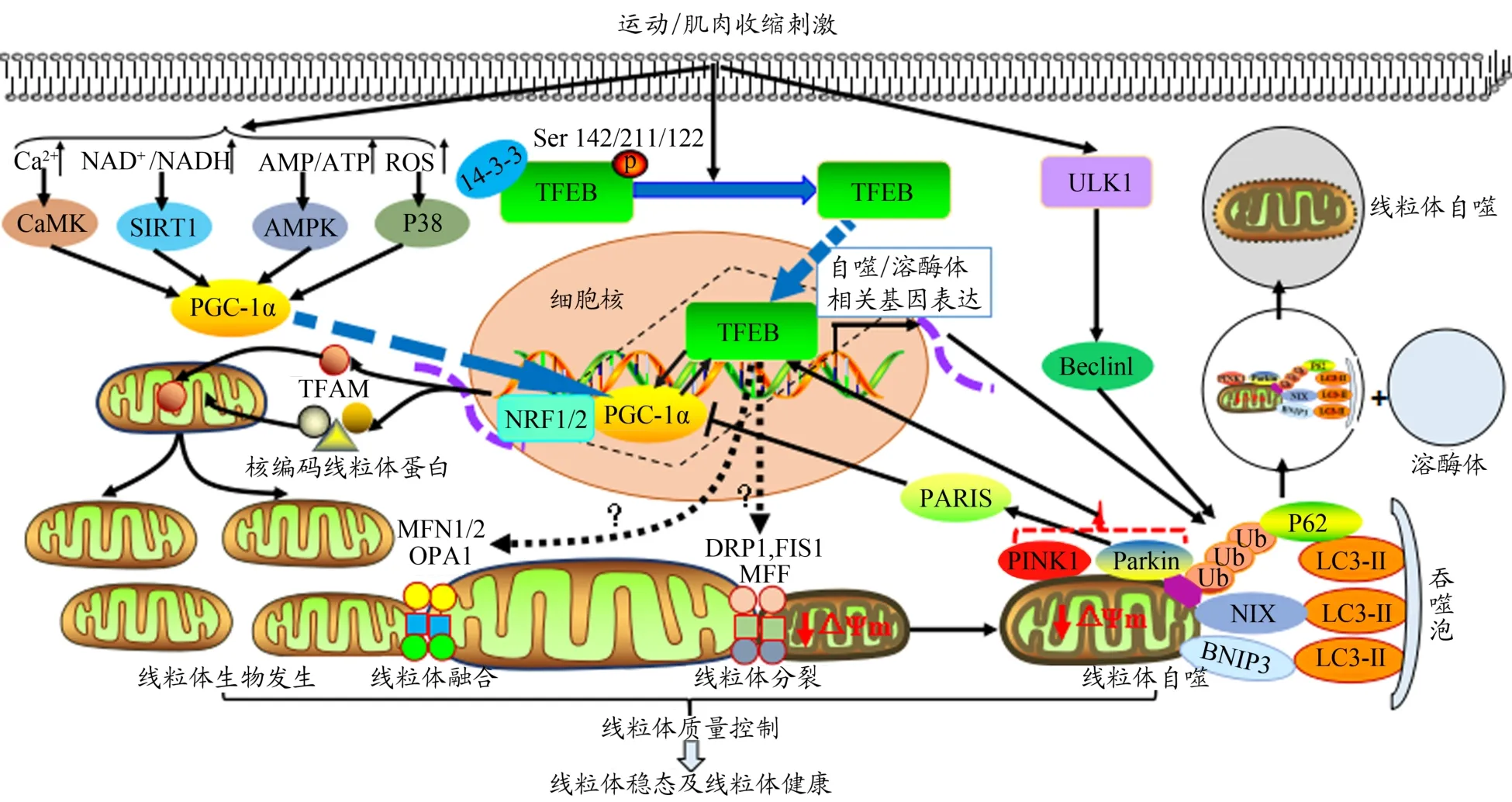
图3 运动/肌肉收缩刺激介导TFEB调节线粒体质量控制及线粒体稳态调控系统的关键内在机制Figure 3.Key Intrinsic Mechanism of Exercise/Muscle Contractile Activity-mediated TFEB in Regulating Mitochondrial Quality Control and Mitochondrial Homeostasis System
目前尚存在一些亟需进一步研究与探讨的重要问题:1)AMPK与SIRT1均是运动介导线粒体质量控制的关键信号分子。据报道,AMPK可通过mTORC1依赖或非依赖两种路径激活TFEB及其转录调节机制(Diogo et al.,2018;Fernandez-Mosquera et al.,2017;Kim et al.,2017a),SIRT1亦可直接去乙酰化激活TFEB及其转录调节机制,进而改善溶酶体活性及功能(Bao et al,2016)。据此,AMPK-SIRT1信号轴可否与TFEB-PGC-1α共调控回路构成信号偶联机制,甚至产生信号级联放大效应,进而在运动介导线粒体质量控制中发挥重要作用亟需明晰;2)TFEB与线粒体融合分裂关键蛋白的具体交互作用及调控关系目前仍未完全明晰,其可否在运动介导的线粒体融合分裂循环中发挥重要作用尚需进一步探讨;3)尽管已有研究认为,PINK1/Parkin可作为TFEB上游的关键信号机制,对线粒体自噬进行特异性控制,但其具体调控机制却知之甚少。基于PINK1具有线粒体丝/苏氨酸蛋白激酶的特性,推断,PINK1或许能直接磷酸化激活TFEB,或通过引发一连串瀑布式的激酶链(kinase cascade),进而间接激活并促进其靶向入核,但这尚需强有力的实验论证;4)当前研究仅证实,TFEB与NIX/BNIP3在运动介导线粒体自噬调控中具有同步变化效应或协同作用特性。但二者在运动介导线粒体自噬及线粒体质量控制中的具体上下游分子调控关系并不明确,有待进一步实验论证;5)FUN14结构域蛋白(FUN14 domain containing 1,FUNDC1)是近期发现的新型线粒体自噬受体蛋白,在运动介导线粒体自噬调控中具有重要作用(Fu et al.,2018)。但FUNDC1与TFEB是否存在具体交互作用及精准上下游分子调控关系,甚至建立信号联动机制,进而在运动介导线粒体自噬及线粒体质量控制中发挥关键作用亦未探明。总之,未来以运动介导TFEB调节线粒体质量控制为关键靶点的系列研究,将会进一步丰富与完善线粒体稳态调控系统的分子信号理论,进而为运动防控慢性代谢疾病提供重要理论参考依据。
