蒙成药小儿清肺八味丸的质量控制方法研究
2020-04-07赵丽娜籍学伟王玉华
赵丽娜 籍学伟 王玉华
【摘 要】 目的:建立蒙成药小儿清肺八味丸的质量控制方法。方法:采用薄层色谱法对小儿清肺八味丸中红花、麦冬、拳参、人工牛黄、胡黄连进行定性鉴别;采用高效液相色谱法测定胡黄连中胡黄连苷Ⅰ和胡黄连苷Ⅱ的含量。结果:薄层鉴别方法专属性强,特征斑点清晰,阴性对照无干扰;胡黄连苷Ⅰ在3.8~76μg/mL范围内与峰面积呈良好线性关系(r=0.9999),平均回收率为99.71%,RSD为1.03%;胡黄连苷Ⅱ在8.4~168μg/mL范围内与峰面积呈良好线性关系(r=0.9999),平均回收率为103.30%,RSD为1.95%。结论:所建立的定性、定量分析方法简便、准确、灵敏度高,可用于小儿清肺八味丸的质量控制。
【关键词】 小儿清肺八味丸;薄层色谱法;高效液相色谱法
【中图分类号】R917 【文献标志码】 A 【文章编号】1007-8517(2020)3-0027-06
Abstract:Objective To establish the quality control method of Xiaoer Qingfei Bawei Pills.Methods Carthami flos、Ophiopogonis radix、Bistortae rhizoma、Bovis calculus artifactus and Picrorhizae rhizoma in Xiaoer Qingfei Bawei Pills were identified by TLC.The contents of Picroside Ⅰ and Picroside Ⅱ in Picrorhizae rhizoma were determined by HPLC.Results The identification method was exclusive.The TLC spots were fairly clear,and the blank test showed no interference.The picroside Ⅰ showed a good linearity in the range of 3.8~76μg/mL(r=0.9999).The average recovery was 99.71%,and RSD was 1.03%.The Picroside Ⅱ showed a good linearity in the range of 8.4~168μg/mL(r=0.9999).The average recovery was 103.30%,and RSD was 1.95%.Conclusion The established qualitative and quantitative methods are simple、accurate and sensitive, which can be used in the quality control of Xiaoer Qingfei Bawei Pills.
Keywords:Xiaoer Qingfei Bawei Pills;TLC;HPLC
蒙成藥小儿清肺八味丸(蒙名:胡勒森竹岗-8)收载于《卫生部药品标准》蒙药分册第63页[1],是蒙医常用儿科用药,由天竺黄、北沙参、红花、胡黄连、牛黄、拳参、檀香、麦冬八味药材制成,具有清肺热、止咳定喘的功效,用于小儿肺热、发烧、咳嗽、气促、瘟疫热盛等症。小儿清肺八味丸现行质量标准只收载了【性状】及【检查】项,无【鉴别】和【含量测定】项,现行标准水平低且不完善。为提高现行质量标准,该实验采用TLC法和HPLC法分别对小儿清肺八味丸薄层色谱鉴别方法和含量测定方法进行研究,并建立红花、麦冬[2]、拳参、人工牛黄、胡黄连[3]等五味药材的薄层鉴别方法及高效液相色谱测定胡黄连化学成分的含量测定方法。
1 仪器与材料
1.1 仪器 戴安U-3000型高效液相色谱仪(美国戴安公司);TLC Visualizer薄层色谱数码成像系统(杭州汉泽科技有限公司);KQ-500DE型超声波清洗器(昆山市超声仪器有限公司);Mettler AE-100型电子天平(万分之一,梅特勒-托利多仪器有限公司);Sartorius ME 5型电子天平(百万分之一,赛多利斯贸易有限公司);DHG-9145A型鼓风干燥箱(上海一恒科学仪器有限公司)。
1.2 药品与试剂 小儿清肺八味丸(乌兰浩特中蒙制药有限公司(A公司),批号:160814、160815、160816,规格:2g /10粒;180403,规格:1g /25粒;内蒙古蒙药股份有限公司(B公司),批号:1608041、1608042、1608043、1712001,规格:1g /25粒;内蒙古库伦蒙药有限公司(C公司),批号:1609071、1609072、1609073、180316,规格:1g /25粒)。
红花对照药材,批号:120907-201412;麦冬对照药材,批号:121013-201310;没食子酸对照品,批号:110831-201605;猪去氧胆酸对照品,批号:100087-201411;胆酸对照品,批号:100078-201415;胡黄连苷Ⅰ对照品,批号:111727-201702,含量:95.6%;胡黄连苷Ⅱ对照品,批号:111596-201805,含量:93.2%。上述对照药材及对照品均来自中国药品生物制品检定研究院。
硅胶G、H、GF254、HSG薄层板(烟台市化学工业研究所、青岛海洋化工厂);德国Merck薄层层析硅胶粉H型(德国默克公司);甲醇为色谱纯,其他试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 薄层色谱鉴别
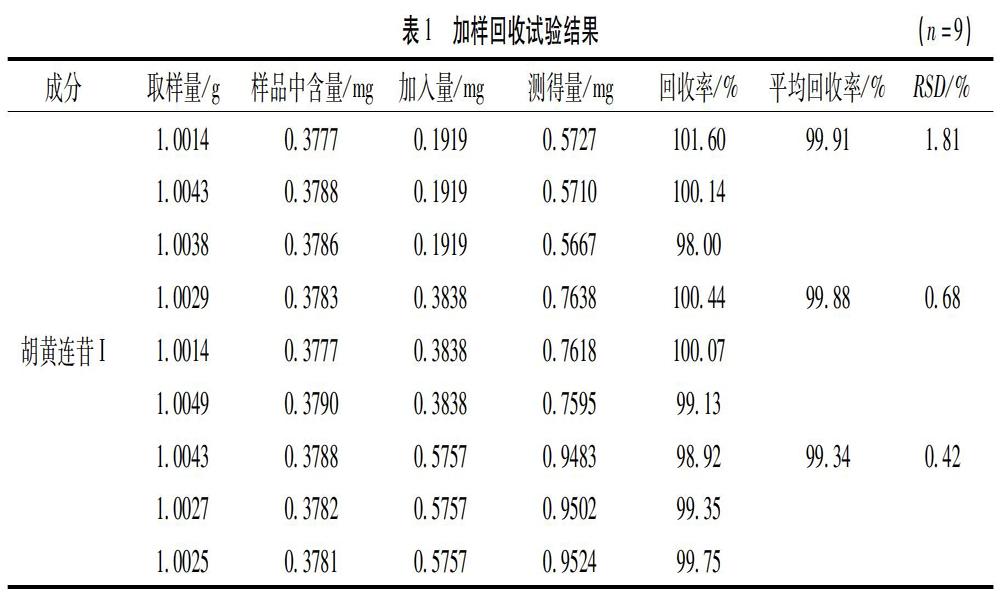
2.1.1 红花 取本品粉末2g,加80%丙酮10mL,密塞,振摇15min,滤过,滤液作为供试品溶液。取红花对照药材约0.5g,同法制成对照药材溶液。另取除红花药材外其他七味药材粉末,按处方比例混合均匀,称取约1.7g,同法制成阴性对照溶液。照薄层色谱法(《中国药典》2015版通则0502)[4]试验,吸取供试品溶液、红花对照药材溶液以及阴性对照溶液各10μL,分别点于同一硅胶H薄层板上,以乙酸乙酯-甲酸-水-甲醇(7∶[KG-*3/5]2∶[KG-*3/5]3∶[KG-*3/5]0.4)为展开剂[5-6],展开,展距为12cm,取出薄层板,晾干。结果显示,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,且阴性对照对测定没有干扰。薄层层析图谱如图1所示。
2.1.2 麦冬 取本品粉末2.5g,加水30mL,再加盐酸2mL,加热回流1h,冷却,滤过,滤液分别用30mL三氯甲烷振摇提取2次,合并三氯甲烷液,蒸干,残渣加三氯甲烷2mL使溶解,作为供试品溶液。取麦冬对照药材0.5g,同法制成对照药材溶液。另取除麦冬药材外其他七味药材粉末,按处方比例混合均匀,称取约2g,同法制成阴性对照溶液。照薄层色谱法(《中国药典》2015版通则0502)试验,吸取供试品溶液与阴性对照溶液各8μL,麦冬对照药材溶液5μL,分别点于同一硅胶G薄层板上,以三氯甲烷-丙酮(4∶[KG-*3/5]1)为展开剂[7],展开,展距为12cm,取出薄层板,晾干,以10%硫酸乙醇溶液为显色剂,均匀喷于薄层板,在105℃加热至斑点显色清晰,在自然光下检视。结果显示,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,且阴性对照对测定没有干扰。薄层层析图谱如图2所示。
2.1.3 拳参 取本品粉末5g,加甲醇20mL,超声处理30min,滤过,滤液蒸干,残渣加甲醇5mL使溶解,作为供试品溶液。取没食子酸对照品适量,加甲醇制成浓度为1mg/mL的溶液,作为对照品溶液。另取除拳参药材外其他七味药材粉末,按处方比例混合均匀,称取约5g,同法制成阴性对照溶液。照薄层色谱法(《中国药典》2015版通则0502)试验,吸取供试品溶液与阴性对照溶液各8μL、没食子酸对照品溶液5μL,分别点于同一硅胶HSG薄层板上,以二氯甲烷-乙酸乙酯-甲酸(5∶[KG-*3/5]4∶[KG-*3/5]1)为展开剂[8],上行展开约12cm,取出薄层板,挥干溶剂后置氨蒸气中熏至斑点显色清晰。结果显示,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的清晰斑点,且阴性对照对测定没有干扰。薄层层析图谱如图3所示。
2.1.4 人工牛黄 取本品粉末1.5g,加甲醇10mL,超声处理20min,摇匀,滤过,取滤液作为供试品溶液。取猪去氧胆酸对照品,加甲醇制成浓度为1mg/mL的溶液,作为对照品溶液。另取除人工牛黄药材外其他七味药材粉末,按处方比例混合均匀,称取约1.5g,按供试品溶液制备方法制成阴性对照溶液。照薄层色谱法(《中国药典》2015版通则0502)试验,吸取供试品溶液与阴性对照溶液各8μL,猪去氧胆酸对照品溶液2μL,分别点于同一硅胶G薄层板上,以正己烷-乙酸乙酯-醋酸-甲醇(20∶25∶2∶3)上层溶液为展开剂[9-10],展开,展距为12cm,取出薄层板,晾干,喷以10%磷钼酸乙醇溶液,待乙醇挥干后,置于烘箱中105℃加熱至斑点显色清晰。结果显示,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,且阴性对照对测定没有干扰。薄层层析图谱如图4。
2.1.5 胡黄连 取小儿清肺八味丸约10g,加甲醇10mL,超声处理20min,滤过,滤液作为供试品溶液。取胡黄连苷Ⅰ对照品、胡黄连苷Ⅱ对照品适量,分别加甲醇制成浓度为1mg/mL的溶液,作为对照品溶液。另取除胡黄连药材外其他七味药材粉末,按处方比例混合均匀,称取约10g,按上述供试品溶液制备方法制备阴性对照溶液。照薄层色谱法(《中国药典》2015版通则0502)试验,吸取供试品溶液、胡黄连苷Ⅰ对照品、胡黄连苷Ⅱ对照品、阴性对照溶液各10μL,分别点于同一硅胶GF254薄层板上,以水饱和乙酸乙酯为展开剂[11],展开,展距为12cm,取出,晾干,再以氯仿-甲醇-乙酸乙酯-甲酸(7∶[KG-*3/5]3∶[KG-*3/5]5∶[KG-*3/5]0.1)为展开剂,展开,取出,晾干,置254nm紫外光灯下检视。结果供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,且阴性对照对测定没有干扰。薄层层析图谱如图5所示。
2.2 含量测定
2.2.1 色谱条件 色谱柱:Agilent Eclipse Plus C18(250mm×4.6mm, 5μm);流动相:甲醇-0.2%磷酸水溶液(31∶[KG-*3/5]69);流速∶[KG-*3/5]1.0mL/min;柱温:30℃;进样量:10μL;检测波长:275nm。
2.2.2 对照品溶液的制备 取胡黄连苷I对照品、胡黄连苷Ⅱ对照品适量,精密称定,加30%乙醇,制成浓度分别为38μg/mL、84μg/mL的胡黄连苷I和胡黄连苷Ⅱ混合对照品溶液,即得。
2.2.3 供试品溶液的制备 精密称定小儿清肺八味丸粉末2g ,于具塞锥形瓶中,精密加入30%乙醇25mL ,密塞,称定重量,超声处理(功率500W,频率40kHz)40min,放冷,再称定重量,用30%乙醇补足减失的重量,摇匀,离心,取续滤液,过微孔滤膜(0.45μm),即得。
2.2.4 阴性对照溶液的制备 按处方比例,制备不含胡黄连的阴性样品,再照“2.2.3”项下操作制备阴性对照溶液,即得。
2.2.5 专属性试验 精密吸取对照品溶液、供试品溶液及阴性对照溶液各10μL,按“2.2.1”项下色谱条件进样并记录色谱图。供试品呈现与对照品保留时间相同的色谱峰,阴性对照无干扰。色谱图如图6所示。
2.2.6 线性关系考察 精密称取胡黄连苷I对照品和胡黄连苷Ⅱ对照品适量,置50mL量瓶中,加入30%乙醇制成胡黄连苷I和胡黄连苷Ⅱ浓度分别为76μg/mL和168μg/mL的混合对照品储备液。分别精密吸取上述储备液0.5、1.5、2.5、5、7.5、10mL,置10mL量瓶中,加30%乙醇稀释至刻度,摇匀。按“2.2.1”项下色谱条件测定峰面积,以峰面积(Y)为纵坐标,对照品浓度(X)为横坐标,进行线性回归,得胡黄连苷I 、胡黄连苷Ⅱ回归方程分别为Y=262.65X+0.030(r=0.9999)、Y=121.76X+0.025(r=0.9999)。结果表明,胡黄连苷I(3.8~76μg/mL)和胡黄连苷Ⅱ(8.4~168μg/mL)均在对应浓度范围内呈良好的线性关系。
2.2.7 稳定性试验 取同一供试品溶液(批号1609071),分别于室温下0、6、10、18、24h测定含量以考察溶液的稳定性。结果显示,在不同时间点测得胡黄连苷I峰面积RSD=0.88%,测得胡黄连苷Ⅱ峰面积RSD=0.19%。表明供试品溶液在24h内稳定。
2.2.8 精密度试验 取同一供试品溶液(批号1609071),连续进样6次,分别测得6次进样的峰面积积分值。结果显示,胡黄连苷I峰面积RSD=0.60%(n=6),胡黄连苷Ⅱ峰面积RSD=0.50%(n=6)。表明仪器精密度好。
2.2.9 重复性试验 取同一供试品(批号1609071)6份,按“2.2.3”项下方法制备供试品溶液,按“2.2.1” 的色谱条件进样并记录色谱图。分别计算胡黄连苷I和胡黄连苷Ⅱ含量。结果显示,胡黄连苷I平均含量为0.38mg/g,RSD为1.06%(n=6);胡黄连苷Ⅱ的平均含量为0.82mg/g,RSD为0.70%(n=6)。表明方法重复性好。
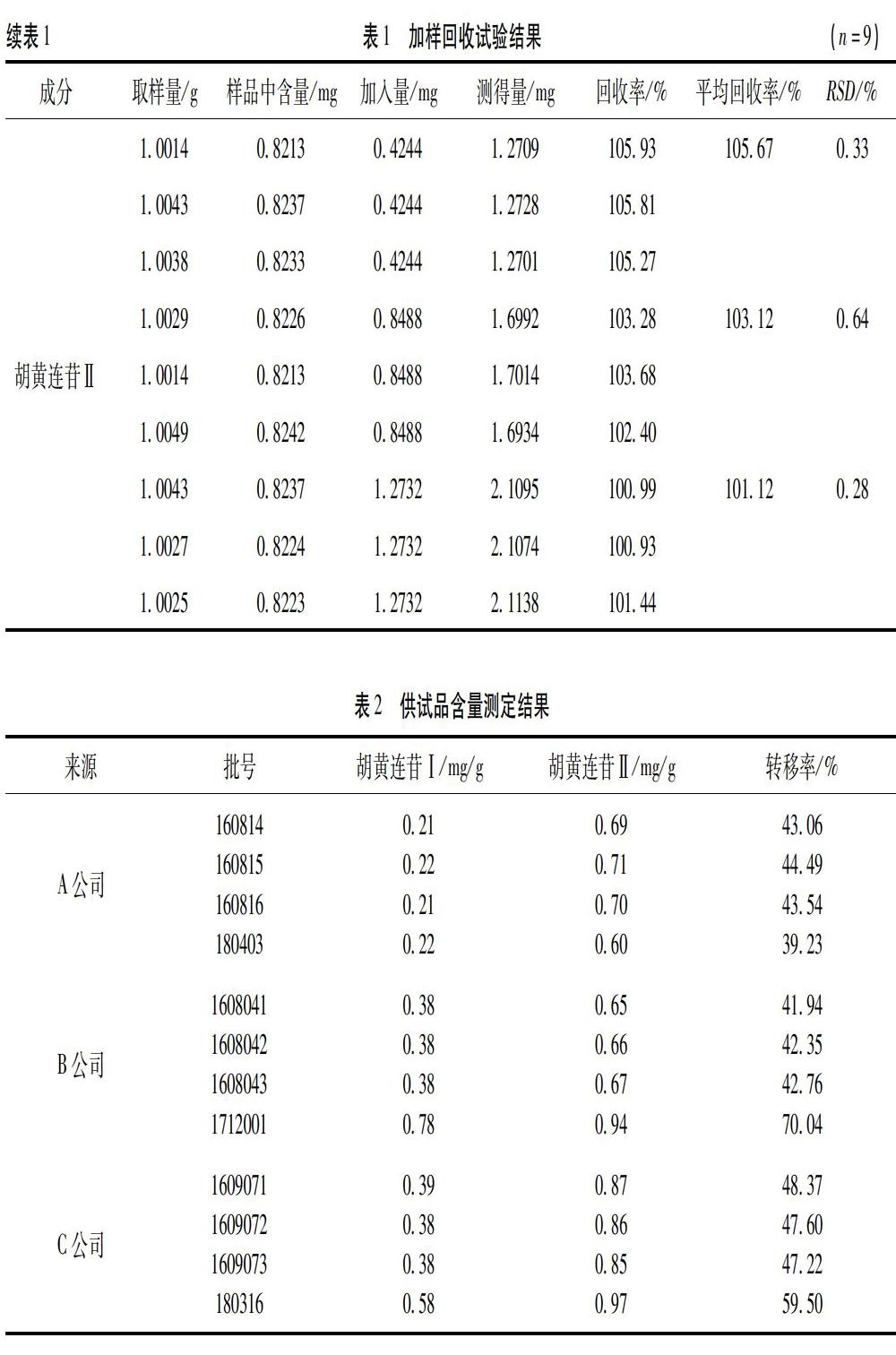
2.2.10 加样回收试验 取已知含量供试品(批号1609071)9份,每份约1g,分别加入一定量的胡黄连苷I和胡黄连苷Ⅱ对照品贮备液,按“2.2.3”項下方法制备供试品溶液,按“2.2.1” 的色谱条件进样并记录色谱图。计算回收率,结果见表1。
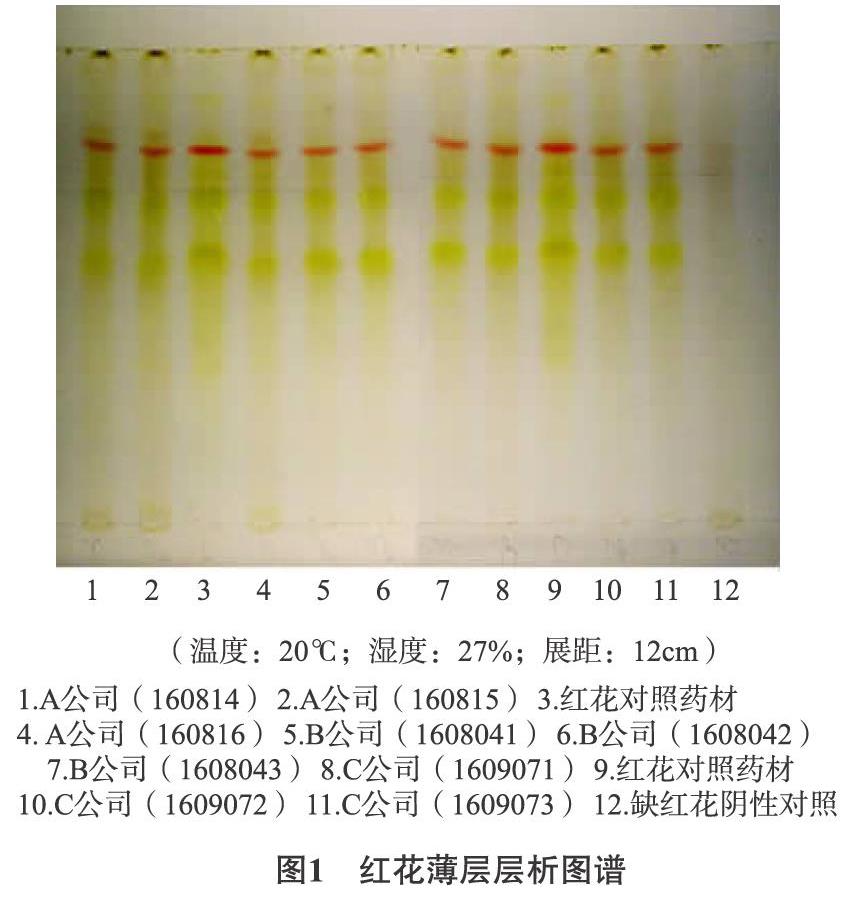
2.2.11 供试品测定 取12批供试品各约2g,按“2.2.3”项下方法制备供试品溶液,按“2.2.1 色谱条件”进样并记录色谱图。计算胡黄连苷I和胡黄连苷Ⅱ的含量。结果见表2。
2.2.12 含量限度的制定 采用HPLC法对12批供试品及3批胡黄连药材进行测定,结果胡黄连苷Ⅰ与胡黄连苷Ⅱ的平均转移率47.51%,由于不同产地胡黄连的胡黄连苷Ⅰ与胡黄连苷Ⅱ含量有所不同,参考《中国药典》2015年版一部“胡黄连”项下有关规定,确定本品每1g含胡黄连苷Ⅰ与胡黄连苷Ⅱ的总量不得少于0.078%。今后可进一步研究积累相关数据再作调整。
3 讨论
3.1 薄层色谱鉴别 在红花的薄层鉴别中,相比青岛海洋化工厂硅胶H板,烟台市化学工业研究所H板斑点清晰、分离度好、Rf值适中;分析原因可能是不同生产企业所用的硅胶质量有差别。为验证方法的耐用性,增加人工手铺薄层板对照试验。结果显示,其与烟台化学工业研究所H板的效果一致。
参照《中国药典》2015年版“人工牛黄”鉴别项下以胆酸、猪去氧胆酸对照品为指标建立小儿清肺八味丸中人工牛黄的薄层色谱鉴别方法。结果在与胆酸对照品相应的位置上,阴性有干扰,在与猪去氧胆酸对照品相应位置上,阴性无干扰。因此本次试验仅建立以猪去氧胆酸为对照品的薄层色谱鉴别方法。
经查阅文献,天竺黄的主要化学成分为硅酸盐、无机元素及氨基酸[12-13],参照《中国药典》2015年版一部“天竺黄”鉴别项下以亮氨酸、丙氨酸为对照品,对小儿清肺八味丸进行薄层色谱鉴别研究[7],结果阴性对照有干扰,方法专属性不强,需要进一步研究。
此外,上述5个薄层色谱鉴别方法均对不同温度(3.5℃、4.1℃、 2.2℃、4.5℃、3.5℃)、湿度(68%、70%、66%、74%、72%)条件进行了验证,结果显示建立的薄层色谱鉴别方法耐用性良好。
3.2 色谱条件选择 参照《中国药典》2015年版一部“胡黄连”项下的含量测定方法[7],以甲醇-水-磷酸溶液(35∶[KG-*3/5]65∶[KG-*3/5]0.1)为流动相进行试验,结果显示,色谱峰分离度较差且胡黄连苷Ⅰ色谱峰有拖尾现象。经试验摸索将流动相调整为甲醇-0.2%磷酸溶液(31∶[KG-*3/5]69)时,色谱峰分离度达到要求、拖尾现象改善、出峰时间适中、峰型较好[14]。
3.3 提取条件选择 胡黄连中胡黄连苷I和胡黄连苷Ⅱ是环烯醚萜类化合物,易溶于水和甲醇,可溶于乙醇、丙酮和正丁醇等[15]。因此试验分别比较了水、甲醇、30%乙醇、70%乙醇、乙醇的提取效果。结果表明,30%乙醇提取效果最佳,峰型最好;试验对回流提取和超声提取进行了比较[16]。结果表明,回流提取与超声提取效率相似。考虑到方法的简便易行,确定采用超声提取方法;试验对提取时间进行了考察。结果表明,超声提取40min供试品中胡黄连苷Ⅰ和胡黄连苷Ⅱ的含量基本稳定不变,故将超声提取时间定为40min。
该实验为小儿清肺八味丸提供了科学、可靠的质量标准,确保了用药的安全、有效。
参考文献
[1]中华人民共和国卫生部药典委员会.中华人民共和国卫生部药品标准·蒙药分册[S].1998:63.
[2]马新换,张丽.咽炎无糖颗粒中药材的定性鉴别研究[J].西部中医药,2018,31(1):28-30.
[3]王锦红,康儿灵颗粒薄层色谱鉴别方法的改进[J].中国药师,2011,4(12):1823-1824.
[4]国家药典委员会.中华人民共和国药典:2015年版四部[S].北京:中国医药科技出版社,2015:57-58.
[5]姚东,包永睿,王帅,等.芦黄参花胶囊质量标准研究[J].中国医药导报,2017,14(1):8-11.
[6]赵超,白晓丹,李锐莉,等.川参胶囊的质量标准提高研究[J].中国药房,2018,29(21):2902-2907.
[7]国家药典委员会.中华人民共和国药典:2015年版一部[S].北京:中国医药科技出版社,2015:1712,56,242-243.
[8]马善波,曹金一,杜群,等.舒乐胶囊质量标准的提高研究[J].中国药师,2017,20(3):446-448,452.
[9]毕力格,柳萌,韩斯古楞.蒙药自拟处方泵嘎日-17的指纹图谱质量标准相关性研究[J].中国民族医药杂志,2017,23(11):79-81.
[10]刘威,张洪娟.丹芪脑血康胶囊制剂工艺及薄层鉴别研究[J].黑龙江中医药,2015,44(4):71-72.
[11]邬国庆,张小茜.胡黄连质量评价方法的研究[J].中草药,2005(12):1886-1888.
[12]王春柳,李晔,张红,等.蒙药天竺黄的研究概况[J].中国药业,2016,25(23):1-5.
[13]曹瑞珍,白靓,张国文,等.蒙药德都红花-7味散各组方的化学成分和现代药理作用研究进展[J].内蒙古民族大学学报(自然科学版),2018,33(1):79-84.
[14]石欣,鲍劲松,白雅静.RP-HPLC法测定清热八味胶囊中胡黄连苷Ⅰ的含量[J].中国民族医药杂志,2011,17(3):52-54.
[15]段晓颖,高卫芳,吴彩丽,等.均匀设计法优选胡黄连中胡黄连苷的提取工艺[J].中国药房,2011,22(39):3675-3676.
[16]马丹凤,李素娟,刘雯.HPLC波长切换法同时测定金衣万应丸中多种有效成分的含量[J].儿科药学杂志,2018,24(6):48-51.
