血管胰岛素抵抗:一个维系心血管和代谢性疾病的潘多拉魔盒
2020-04-07田怡周明生
田怡,周明生
(1.西北农林科技大学资源环境学院,陕西 杨凌 712100;2.沈阳医学院基础医学院生理学教研室)
胰岛素抵抗是指各种原因引起的胰岛素调节组织葡萄糖代谢功能降低的状况。由于骨骼肌、肝脏和脂肪组织是胰岛素调节糖和脂肪代谢的主要脏器,我们通常所指的胰岛素抵抗是由于胰岛素信号通路在这些传统的代谢组织受损引起的机体代谢功能紊乱[1]。胰岛素抵抗不仅发生在一些传统的胰岛素代谢组织,而且也发生在心血管、免疫细胞和其他各种组织。我们常常把损害血管胰岛素信号通路和舒张功能这种状态称为血管胰岛素抵抗[2-3]。
在血管,胰岛素除调节代谢外,还激活2条信号通路[3],一是激活血管内皮细胞磷脂酰肌醇-3-激酶 (PI3K) /Akt蛋白激酶通路,促进内皮型一氧化氮合成酶 (eNOS)丝氨酸位点磷酸化增加一氧化氮 (NO)产生,引起血管扩张;二是激活有丝分裂原活化蛋白激酶 (MAPK)引起血管收缩和血管平滑肌细胞 (VMSC)增殖[2]。在生理状况下,胰岛素激活PI3K/Akt/NO信号通路,引起血管扩张,维持血管内环境稳定,对心血管系统起保护作用。当发生血管胰岛素抵抗时,胰岛素激活PI3K/NO通路选择性受损,代偿性高胰岛素血症激活MAPK引起VMSC增殖、血管肥厚,促进高血压和心血管疾病的发生[2-3]。由于失去胰岛素扩血管效应,使骨骼肌等代谢组织的血流量减少,降低葡萄糖摄取,血管胰岛素抵抗有增加代谢性胰岛素抵抗的风险[4]。胰岛素抵抗是代谢综合征发生的主要原因,在临床上,胰岛素抵抗常与肥胖、高血压、血管内皮细胞功能损害以及一些其他的心血管危险因素关联在一起,共同促进心血管疾病和糖尿病的发生[5-6]。由于血管胰岛素信号通路在维护血管内环境稳定及调节糖代谢中的重要作用,血管胰岛素抵抗可能是心血管和代谢性疾病相互依存、相互共生的病理基础,以及维系心血管和代谢性疾病的一个重要纽带[3]。本文结合实验性和临床数据,阐明了胰岛素在心血管系统中的生理和病理生理作用以及在促进心血管和代谢疾病发展的作用和机制。
1 胰岛素的心血管作用
众所周知,胰岛素是一种促进合成代谢激素,在调节肝脏、脂肪组织和骨骼肌的葡萄糖和脂质代谢作用中起着重要作用。除了调节代谢,胰岛素对心血管系统具有复杂的调节作用[3,7]。一方面,胰岛素诱导血管内皮NO生成促进血管舒张,抑制VSMC迁移和增殖,抑制炎症和血栓形成,产生血管保护效应;另一方面,在一些病理性情况下,胰岛素通过激活MAPK信号通路引起血管收缩和促动脉粥样硬化发生,从而损害心血管功能[4,7]。胰岛素对心血管系统的作用与循环血液中的胰岛素浓度和病理生理状况有密切关系。在生理状况下,胰岛素通过刺激NO生成对心血管产生保护效应;而在病理状况下,高胰岛素血症可能引起血管损伤[2](图 1)。
1.1 胰岛素的扩血管效应 研究表明胰岛素可引起动静脉血管舒张并增加微循环流量增加。在正血糖钳夹实验时胰岛素灌注可剂量依赖性地增加骨骼肌血流量[8]。此外,胰岛素对各种血管床的扩张效应也会随着时间和剂量的不同而产生差异。胰岛素灌注早期引起末梢小动脉的扩张并增加灌注毛细血管的数量,导致毛细血管募集,然后扩张直径较大的阻力血管,从而增加总的肢体血液[8]。研究表明胰岛素的血管扩张作用主要是由于刺激血管内皮细胞释放 NO所致[8-9]。在骨骼肌,胰岛素的舒血管效应有助于骨骼肌摄取葡萄糖并进行代谢[10]。正常状况下,机体70%的葡萄糖都在骨骼肌细胞进行代谢,因此,胰岛素的扩血管效应除直接参与血流动力学调节外,在糖代谢中也起到非常重要的作用[3]。
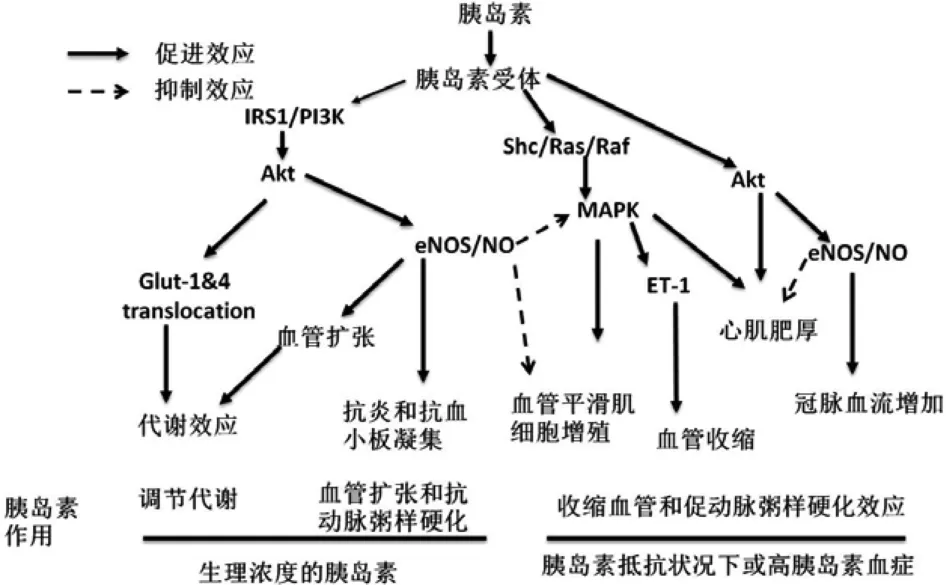
图1 胰岛素的心血管效应
1.2 胰岛素的缩血管效应 在血管系统,除刺激NO诱导血管舒张,胰岛素还刺激交感神经以及血管内皮细胞释放内皮素 (ET)-1,引起血管收缩[11-13]。胰岛素灌注可增加健康志愿者血浆儿茶酚胺浓度和骨骼肌交感神经活性,给狗颈动脉灌注生理浓度的胰岛素可增加动脉血压,该效应可通过阻断神经节而废除,表明该效应是胰岛素激活交感神经所致[13]。由于MAPK是调节血管内皮细胞ET-1释放的上游信号分子,因此,在PI3K信号通路受损时,胰岛素可能激活MAPK/ET1通路引起血管收缩[11]。此外,胰岛素对骨骼肌小动脉的收缩效应还可被MAPK抑制剂所逆转。在生理状况下,胰岛素引起的NO扩血管效应常常占主导地位,胰岛素缩血管效应常因其扩血管效应而被忽略。然而,在胰岛素抵抗状态,例如在肥胖和糖尿病时,胰岛素NO扩血管效应受到损害,胰岛素缩血管效应可能得到增强[11,14],这也可能是肥胖和糖尿病等胰岛素抵抗代谢性疾病引起血压增加或易发生高血压的一个原因。
1.3 胰岛素的抗炎和抗动脉粥样硬化效应 研究表明生理浓度的胰岛素具有抗炎和抗动脉粥样硬化的作用,其机制可能涉及血管内皮NO的产生。NO是一个重要的心血管保护分子,NO引起血管舒张并抑制VSMC的生长和增殖[2,7]。血管内皮是胰岛素分泌入循环血中后遇到的第一个脏器。在生理状况下,循环血中保持着一定浓度的胰岛素水平,胰岛素可持续地刺激内皮细胞产生NO并维持血管壁上NO水平,起到维护心血管内环境稳定的作用。研究发现生理浓度的胰岛素能抑制血管内皮转录核因子 (NF)-κB及单核细胞趋化蛋白(MCP)-1 mRNA表达,这些作用是通过NO依赖性途径[15]。此外,文献报道给肥胖者静脉滴注低剂量胰岛素和5%葡萄糖可明显减少氧自由基(ROS)生成和一些可溶性细胞间黏附分子(ICAM) -1 和 MCP-1 生成[16]。 在 ApoE-/-动脉粥样硬化易发模型小鼠,胰岛素治疗可减少动脉粥样硬化斑块的数量和大小[17]。低剂量胰岛素治疗明显改善急性心肌梗死患者临床预后[18]。这些结果支持胰岛素具有抗炎和抗动脉粥样硬化作用。
1.4 胰岛素的致炎和促动脉粥样硬化效应 胰岛素是一种促进组织细胞合成和生长的生长因子,可调节多种原癌基因转录因子的活性,包括c-fos和MAPK。在培养的VSMC中,胰岛素刺激VSMC增殖和迁移,这些作用主要通过激活MAPK途径[19]。胰岛素样生长因子1、胰岛素原和胰岛素均可刺激血管生长和促进动脉粥样硬化形成。流行病学研究表明与其他人群相比,肥胖和2型糖尿病患者患动脉粥样硬化的风险明显增加且伴有高胰岛素血症[20]。动物实验和临床研究也表明因胰岛素抵抗引起的代偿性高胰岛素血症可增加血管内皮血浆纤溶酶原激活物抑制剂-1(PAI-1)、细胞黏附分子 (VCAM)-1和E-选择素的表达,加速动脉粥样硬化产生[21]。因此,肥胖和糖尿病患者易患动脉粥样硬化可能与高胰岛素血症促进动脉粥样硬化的效应有关。
2 血管胰岛素信号通路
2.1 血管内皮细胞胰岛素信号通路 在血管内皮细胞,胰岛素刺激2条主要信号通路:PI3K和MAPK。胰岛素通过激活PI3K通路产生NO导致血管舒张并增加血流[3]。胰岛素调节内皮NO产生的信号通路业已阐明:该反应始于循环中的胰岛素与其受体结合从而启动下游胰岛素信号分子反应[2]。胰岛素受体是一个含有酪氨酸激酶的异四聚体,与胰岛素结合后,引起酪氨酸快速磷酸化,进一步激活胰岛素受体酪氨酸激酶并与胰岛素受体底物 (IRS) 相互作用[22]。IRS包括3个亚型:IRS-1、IRS-2和IRS-3。IRS-1是胰岛素受体酪氨酸激酶的一个主要底物,也是胰岛素在内皮细胞激活PI3K、产生NO的主要中间信号分子[23]。酪氨酸位点磷酸化IRS-1与PI3K的p85调节亚基结合,引起PI3K激活,后者相继促发内皮细胞Akt丝氨酸473位点磷酸化和一氧化氮合酶(eNOS)丝氨酸1177位点的磷酸化,最终增加eNOS 活性和 NO 产生[22-23]。
在血管内皮中,胰岛素NO信号通路与激活G蛋白偶联受体的经典钙依赖性信号通路如乙酰胆碱完全不同,因为用钙螯合剂BAPTA预处理细胞不能抑制胰岛素刺激 eNOS磷酸化和eNOS活性[24]。有研究表明老年大鼠伴有胰岛素血管舒张功能受损,其机制主要与胰岛素介导的Akt/eNOS通路活化有关,然而,在这些老年大鼠乙酰胆碱介导的Ca2+-钙调素/eNOS信号通路并没有受到影响[25]。这些结果支持胰岛素与乙酰胆碱是通过2条不同的信号通路调节eNOS活性和NO产生。
除了eNOS/NO通路,胰岛素还刺激血管内皮的MAPK通路,MAPK参与细胞生长、迁移、致血栓形成和纤维化因子的产生等的调节。胰岛素激活MAPK还促进ET-1分泌和一些黏附分子蛋白 (例如VCAM-1、MCP-1和E-选择素)的表达, 而 PI3K 不参与这些反应过程的调节[2,4,22]。有趣的是,激活MAPK和PI3K信号通路所需的胰岛素水平是不同的。正常人空腹血浆胰岛素水平通常为50~150 picomolar,在此范围内,胰岛素激活PI3K途径,参与调节糖代谢并通过内皮细胞释放NO维持血管张力[3]。在胰岛素抵抗状态,胰岛素刺激的PI3K途径常常选择性受损,而代偿性的高胰岛素血症 (血浆胰岛素水平在数百picomolar以上)可激活MAPK,使胰岛素从血管的保护效应切换到对血管损害效应,因而增加高血压和心血管病的发生[3,22]。因此,血浆胰岛素水平可能是调控胰岛素PI3K和MAPK信号通路平衡的一个重要因素 (图1)。
2.2 VSMC中的胰岛素信号通路 胰岛素受体在人、鼠和牛VSMC中均有表达。VSMC中的胰岛素信号通路与血管内皮细胞中的相似:胰岛素与胰岛素受体结合,导致酪氨酸磷酸化,随后激活下游分子PI3K和Akt的磷酸化。然而,与血管内皮细胞不同,胰岛素在VSMC中激活PI3K/Akt通路增加诱导型 NO合酶 (iNOS)产生 NO而不是eNOS[26]。尽管iNOS生成的 NO也可引起血管扩张,但iNOS的激活常常受炎症细胞因子的调节。胰岛素在血管平滑肌激活PI3K/iNOS通路对血管张力和功能作用还不清楚,有待于进一步明确。除了iNOS,在VMSC,胰岛素激活MAPK并与其他一些促动脉粥样硬化生长因子,如血管紧张素Ⅱ (AngⅡ)相互作用促进动脉粥样硬化过程[3,22]。 Ang Ⅱ及其 1 型受体 (AT1R) 有明显地致炎和促动脉粥样硬化效应。在培养VSMC,胰岛素通过AT1R激活MAPK途径刺激血管紧张素原的表达。另一方面,AngⅡ通过增加IRS1的丝氨酸磷酸化来抑制胰岛素信号通路,因为IRS-1丝氨酸位点磷酸化可抑制酪氨酸位点磷酸化从而阻断胰岛素激活PI3K信号通路[23]。在胰岛素抵抗状态下,过度激活肾素-血管紧张素-醛固酮系统可能损害胰岛素PI3K途径;而AngⅡ和代偿性高胰岛素血症可能协同刺激MAPK,促进心血管疾病[3,22]。
3 血管胰岛素抵抗与心血管和代谢性疾病
心血管疾病、肥胖和糖尿病等代谢性疾病常有胰岛素扩血管功能障碍和胰岛素刺激PI3K/NO信号通路的受损[2,14]。血管胰岛素抵抗可加剧内皮功能障碍和血管扩张功能损害,增加微血管疾变 (如糖尿病视网膜病变和肾病)、血管炎症和动脉粥样硬化的形成[3]。因此,血管胰岛素抵抗可促进心血管疾病的发生发展。由于胰岛素激活PI3K信号通路能促进骨骼肌、脂肪组织中的葡萄糖转运蛋白GLUT-4向细胞膜转运,增加葡萄糖摄取。在肥胖和糖尿病等代谢性疾病,血管胰岛素抵抗除促进血管并发症外,还可能因降低这些代谢组织的葡萄糖摄取而加剧代谢性胰岛素抵抗[4]。
胰岛素激活PI3K通路在血管组织选择性受损害在心血管疾病的发展中具有重要的病理生理意义。在血管内皮细胞,PI3K通路为胰岛素激活eNOS活性所必需,同时,PI3K也是胰岛素调节脂肪组织和骨骼肌糖和脂肪代谢的一个关键信号分子[2]。因此,胰岛素PI3K信号通路同时调节血管和代谢功能可能为临床上解释心血管疾病和胰岛素抵抗代谢性疾病频繁关联性提供了分子生物学基础[2]。
3.1 血管胰岛素抵抗与高血压 高血压,特别是盐敏感性高血压,常伴有胰岛素抵抗包括血管胰岛素抵抗,高血压患者患糖尿病风险也明显增加[27]。动物实验研究表明在多种高血压模型鼠中有胰岛素激活血管内皮细胞PI3K/NO信号通路以及胰岛素诱导的扩血管功能受损[4,28]。在 PI3K/NO通路受损时胰岛素常引起血管收缩而不是扩张效应[2,11]。 由于血管内皮细胞胰岛素 PI3K/NO 在调节代谢和血管功能方面的重要作用,血管胰岛素抵抗可能是高血压增加心血管并发症和代谢性疾病的一个重要机制[4]。目前,高血压引起血管胰岛素抵抗的机制还不清楚。高血压患者常有过度激活的肾素-血管紧张素-醛固酮系统,在培养的血管内皮细胞中,AngⅡ可以在多个水平上干扰胰岛素PI3K信号通路,如AngⅡ可刺激血管内皮细胞IRS1在丝氨酸2个位点 (Ser616位点和Ser312位点)磷酸化,从而抑制胰岛素刺激IRS酪氨酸磷酸化抑制胰岛素通路[23]。我们的研究也发现Dahl盐敏感性高血压大鼠不但有明显的血管损害和代谢性胰岛素抵抗[28-29],而且胰岛素刺激PI3K/eNOS/NO通路明显受损伴有血管AT1R上调和ROS产生增加[29]。AT1R阻断剂或抗ROS治疗在减轻血管损害的同时也改善血管胰岛素信号通路和胰岛素的舒血管功能,提示高血压引起血管胰岛素抵抗可能与过度AngⅡ/ROS有关[29-30]。
3.2 血管胰岛素抵抗与动脉粥样硬化 在生理状态下,胰岛素PI3K/Akt/NO信号通路有助于维持心血管内环境的稳定,具有抗动脉粥样硬化作用[2]。内皮细胞中的NO和Akt都有抗动脉粥样硬化的作用[31],内皮细胞中Akt的缺失会增加动脉粥样硬化发生。研究发现一些促动脉粥样硬化的病理性刺激如AngⅡ、ROS或各种炎症细胞因子均通过诱导内皮细胞IRS1丝氨酸磷酸化而抑制内皮细胞胰岛素通路[22]。糖尿病或其他一些胰岛素抵抗性代谢性疾病,由于失去胰岛素PI3K/Akt/NO生化级联反应通路的抗动脉粥样硬化效应,这些疾病常常增加动脉粥样硬化风险[2]。研究发现在特异性敲除血管内皮胰岛素受体 (EIRAKO)的ApoE-/-小鼠,在胰岛素敏感性、葡萄糖耐量、血浆脂质或血压方面,EIRAKO的小鼠与野生型小鼠没有区别[32]。然而,在EIRAKO小鼠中,动脉粥样硬化病变的严重程度高出2倍以上。动脉粥样硬化增加的机制可能与损失胰岛素PI3K/Akt抑制VCAM-1 作用有关[2,32]。 在 EIRAKO 小鼠中,VCAM-1在动脉血管中的表达增加,这可能导致增加单核细胞的结合和摄取,从而增加了动脉粥样硬化斑块中的炎性细胞[32-33]。因此,在生理状态上胰岛素总体效应表现为抗动脉粥样硬化作用,在糖尿病或胰岛素抵抗状态下改善血管内皮的胰岛素作用可能有助于减少动脉粥样硬化[2]。
3.3 血管胰岛素抵抗与肥胖和糖尿病 随着肥胖等代谢性疾病的流行,肥胖和糖尿病等代谢性疾病已成为心血管疾病最主要危险因素[34]。肥胖和糖尿病不但降低代谢组织的胰岛素敏感性,而且胰岛素刺激血管内皮细胞PI3K/NO通路以及胰岛素诱导的血管扩张效应也明显受损[4],血管组织选择性的PI3K/NO受损被认为是肥胖等代谢性疾病促进高血压和心血管疾病的重要病理生理学基础。肥胖等代谢性疾病引起血管胰岛素抵抗的机制还未十分清楚,从脂肪组织释放到血液中自由脂肪酸、炎症细胞因子以及各种脂肪素 (adipokines) 都具有重要的血管生物学活性[1,35], 可引起血管内皮细胞IRS1丝氨酸磷酸化而抑制血管胰岛素信号通路。我们最近研究表明从脂肪组织分泌的抵抗素 (resistin)可通过诱导血管内皮细胞的ROS和内质网应激而抑制胰岛素刺激人脐静脉内皮细胞 (HUEVCs)的胰岛素信号通路和胰岛素的血管舒张效应,表明肥胖引起的脂肪素的失平衡可能是引起血管胰岛素抵抗的一个重要机制[36]。
4 结论
肥胖和糖尿病等胰岛素抵抗性代谢性疾病正在成为最主要的心血管病危险因素。在临床上,高血压、肥胖和心血管疾病常常相互并存和相互促进,并以胰岛素抵抗为核心,同时伴有血管内皮细胞功能障碍等众多的心血管功能和结构的异常[22,37]。 血管胰岛素 PI3K/NO 通路具有对代谢和心血管系统双重的调节效应,在高血压、肥胖以及众多的代谢和心血管疾病都发现有血管胰岛素PI3K/NO通路受损。因此,选择性的损害血管胰岛素通路可能是胰岛素抵抗性代谢性疾病和心血管疾病一个重要的病理生理机制以及维系心血管和代谢性疾病的重要纽带。阐明在这些疾病中血管胰岛素抵抗的机制对防治心血管和代谢性疾病具有十分重要的意义。
