液相色谱-串联质谱法检测肝癌细胞N6-甲基腺嘌呤核苷甲基化水平
2020-04-06李盛建李群英须秋萍周艳卿张国庆
李盛建,王 慧,吕 磊,李群英,须秋萍,周艳卿,张国庆,赵 亮
(1. 上海宝山区罗店医院药剂科,上海 201908;2. 上海东方肝胆外科医院药剂科,上海 200438)
表观遗传,是指在DNA碱基序列不发生变化,其表型或基因表达发生了可稳定遗传的变化,包括DNA甲基化修饰、RNA甲基化修饰及非编码RNA等。目前研究中,DNA甲基化修饰为较清楚的表观遗传学修饰,其影响基因表达、蛋白功能等重要生命过程;同时,转录后多达100多种RNA修饰被认为丰富了RNA功能和遗传多样性,N6-甲基腺嘌呤核苷(N6-methyladenosine,m6A)和5-甲基胞嘧啶核苷是其中最具有代表性的甲基化修饰,而m6A更为普遍,在RNA甲基化修饰中超过80%[1]。近年来,m6A相关的研究涉及RNA翻译效率[2]、DNA损伤修复[3]、生长发育[4]、肿瘤形成[5]等等,可见其重要性。
由于液相色谱-串联质谱法(liquid chromatography-tandem mass spectrometry,LC-MS/MS)具有快速、灵敏、准确性高、检测限低等特点,其应用在表观遗传领域中渐渐崭露头角,如基因组羟甲基胞嘧啶和5-甲基胞嘧啶修饰等研究[6-7],但m6A甲基化修饰的LC-MS/MS分析方法国内外鲜有报道。本研究通过建立一种稳定可靠的LC-MS/MS法测定腺嘌呤核苷(adenosine,A)和m6A含量,分析肝癌细胞株HepG2的m6A甲基化水平,并与正常肝细胞株L02进行比较。
1 材料与方法
1.1 实验细胞 本研究采用的细胞为人源肝癌细胞株HepG2和正常肝组织细胞株L02,均由上海东方肝胆外科医院信号转导实验室惠赠。
1.2 仪器和试剂
1.2.1 仪器:Agilent 1290 UPLC二元液相色谱系统, Agilent 6470 Triple Quad质谱仪(美国Agilent);METTLER AE 240型电子天平(瑞士METTLER TOLEDO);低温离心机(美国Thermo Fisher);Millipore超纯水仪(美国Millipore)。
1.2.2 试剂:对照品腺嘌呤核苷和对乙酰氨基酚(中国食品药品检定研究院),N6-甲基腺嘌呤核苷(美国SELLECK CHEMICALS),Trizol,Ambion®Dynabeads mRNA Purification Kit,mRNA RiboMinusTMHuman Transcriptome Isolation Kit(美国Thermo Fisher),甲醇、乙腈为色谱纯(美国Honeywell),甲酸为色谱纯(美国SIGMA),水为超纯去离子水,其他试剂均为分析纯。
1.3 方法
1.3.1 mRNA的提取和酶解:取对数期生长的HepG2和L02细胞,细胞数量(1~5)×106个;按TRI Reagent®试剂说明书步骤,采用Trizol法提取总RNA;按照Ambion® Dynabeads mRNA Purification Kit和RiboMinusTMHuman Transcriptome Isolation Kit试剂盒说明书中的步骤从总RNA中分离纯化mRNA及去除rRNA;用Nanodrop测定mRNA浓度。取200 ng mRNA,2.5 μl 缓冲盐溶液(10×,20 mmol/L ZnCl2,100 mmol/L NaCl)和1 μl核酸酶至EP管,加双蒸水补齐至25 μl,37°C下反应2h;反应体系中再加入2.5 μl酶缓冲溶液和1 μl碱性去磷酸酶,37°C下反应2h;在反应体系中加入去离子水稀释至200 μl,-80℃冰箱保存待检测。
1.3.2 溶液的配制:分别精密称A,m6A及对乙酰氨基酚对照品10 mg,置于10 ml 容量瓶中,加甲醇溶解,配成1.00 mg/ml对照品储备液;用甲醇按比例逐级稀释配制不同浓度工作液,其中A,m6A混合对照品溶液浓度分别为30.00,100.00,200.00,400.00,2 000.00,10 000.00 ng/ml和3.00,10.00,20.00,40.00,200.00,1 000.00 ng/ml,质控储备液浓度分别为40.00,4 000.00,8 000.00 ng/ml和4.00,400.00,800.00 ng/ml,对乙酰氨基酚内标溶液浓度为20.00 ng/ml。上述对照品储备液置4℃冰箱保存。
分别取10 μl上述对照品工作液,加到190 μl含有酶及酶解缓冲液的空白水基质中,配成m6A与A系列浓度工作液样品。A的质量浓度为1.50,5.00,10.00,20.00,100.00和500.00 ng/ml,随行质控样品低、中、高质量浓度分别为2.00,200.00和400.00 ng/ml;m6A的质量浓度为0.15,0.50,1.00,2.00,10.00,和50.00 ng/ml;低、中、高质量浓度质控样品分别为0.20,20.00和40.00 ng/ml。
1.3.3 色谱及质谱条件:色谱条件:色谱柱为美国Agilent Proshell 120 EC-C18(3.0×100 mm,2.7 m),预柱:Agilent UPLC Guard SB C18(3.0×5 mm,2.7μm)柱温:30℃。流动相系统:水(0.1%FA)∶甲醇=82∶18(v/v)等度洗脱,分析时间3 min,进样量2 μl。质谱条件:采用AJS ESI正离子模式,ESI源参数设置:干燥气温350℃;干燥气流速10 L/min;雾化器压力40 psi;鞘气温度350℃;鞘气流速11 L/min;毛细管电压4 000 V;喷嘴电压2 000 V。动态多反应监测模式参数设置:A检测参数268.0→136.0,碎片电压90V,碰撞能量15 eV;m6A检测参数282.0→150.0,碎片电压70V,碰撞能量15 eV;内标检测参数152.3→110.0,碎片电压110V,碰撞能量17 eV。
1.3.4 待测样品前处理:取待测细胞样品30 μl于1.5 ml EP管中,加入60 μl对乙酰氨基酚内标 (10 ng/ml ),涡旋30 s,4 ℃条件下12 000 g离心5 min;取上清液于进样瓶中,用于LC-MS/MS检测。
2 结果
2.1 LC-MS/MS 方法学验证
2.1.1 方法专属性: 分别取30 μl空白基质、对照品溶液和待测细胞样品,按“1.3.4”项步骤处理(处理空白基质时,内标用甲醇代替),按“1.3.3”项条件下进样分析(见图1),A保留时间为1.3min, m6A保留时间为1.4 min,内标保留时间为2.4 min,对照品溶液和待测细胞样品中待测分析物出峰时间一致,无杂质干扰峰,峰形良好。结果表明,该方法具有良好的专属性。
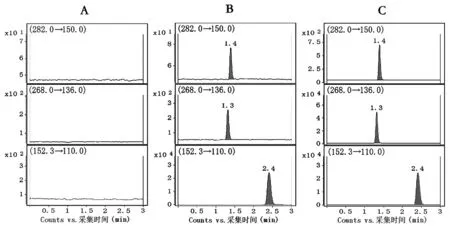
A.空白基质,B.低浓度质控样品,C.待测样品图1 待测物与内标多反应监测色谱图
2.1.2 标准曲线范围与定量下限: 分别取“1.3.2”项中配制好的对照品溶液30 μl,按“1.3.4”项操作,分别配成6个浓度的标准样品,每浓度点配制5份样品,连续进样分析,记录色谱图,以浓度 (X)为横坐标,各待测物的峰面积与内标的比值(Y)为纵坐标进行回归计算,权重系数1/x2,求得回归方程A:Y= 0.040 6X- 0.035 9,r=0.999 9,线性范围为1.50 ~ 500.00 ng/ml;m6A:Y= 0.010 3X- 0.000 9,r= 0.9999,线性范围为0.15 ~50.00 ng/ml。A和m6A分别在1.50 ~500.00 ng/ml和 0.15~50.00 ng/ml范围内具有良好的线性关系,定量限分别为1.50 ng/ml和0.15 ng/ml。
2.1.3 精密度和准确度实验: 按“1.3.2”和“1.3.4”项中的方法制备低、中、高三种浓度的质控样品,每个浓度平行操作5份,进样分析,连续3 天,将被测物和内标的峰面积比值代入标准曲线方程计算实际样品浓度,精密度用相对标准偏差(RSD)表示,结果见表1。m6A日内精密度RSD<13.77 %,日间精密度RSD<11.50 %。A日内精密度RSD<7.84%,日间精密度RSD<5.78%。准确度以相对回收率表示,实测浓度与理论加入浓度的比值即为相对回收率,结果m6A日内、日间准确度范围在93.67 %~101.07 %。A日间、日内准确度范围98.58%~101.10%。提示该方法的精密度和准确度均能达到检测要求。

表1 待测物日间、日内的精密度与准确度(n=5)
2.1.4 基质效应和提取回收率:按“1.3.2”和“1.3.4”项中的方法制备含待测物低、中、高浓度的质控样品;按“1.3.5”项方法处理空白基质(内标用甲醇代替),取上清溶液,加入一定量质控浓度对照品储备溶液,使其最终浓度分别与质控样品的进样浓度一致;配制待测物的甲醇溶液,其最终浓度分别与质控样品的进样浓度一致。进样得峰面积,计算含基质与不含基质样品的峰面积之比,得到相应物质的基质效应;计算基质存在下,前处理和后处理加标样本的峰面积比值,得到待测物的提取回收率。考察内标的基质效应和提取回收率与上述操作一致。结果低、中、高三种浓度待测物及内标提取回收率均在91.46 %~97.60 %之间,基质效应均在90.26 %~99.27 %之间(见表2)。结果表明回收率满足定量要求,基质效应对待测物的定量不产生明显的影响。
2.1.5 稳定性考查:按“1.3.2”和“1.3.4”项中的方法制备含待测物低、中、高浓度的质控样品,考察按以下方式处理后样品的稳定性:室温放置6 h后处理;处理后室温放置24 h;三个冻融循环;-80℃保存30天。测定样品实际浓度并计算RSD和相对偏差(RE),计算公式:RE %=(实测值-真实值)/ 真实值×100 %。结果RSD%均小于15.0 %,RE的范围为0.4 % ~ 6.9 %,表明样品在上述处理状态下稳定性良好。
表2 待测物及内标提取的回收率和基质效应
注:IS: Internal Standard
2.2 样品分析 取“1.3.1”项的HepG2和L02待测样品,按“1.3.4”项处理进样分析。按内标法计算样品中m6A和A的浓度,并根据下列公式计算mRNA中m6A甲基化百分比:m6A %=(Cm6A/ Mm6A)/(CA/MA+Cm6A/Mm6A),Mm6A和MA分别为m6A和A的摩尔质量,Cm6A和CA分别为m6A和A的样品实测浓度。对m6A甲基化水平进行统计分析,差异有统计学意义(P<0.05),表明肝癌细胞株HepG2的m6A甲基化水平高于正常肝细胞株L02。
表3 细胞株HepG2和L02的m6A甲基化水平
3 讨论
目前传统的m6A甲基化分析技术主要有斑点杂交法[8]、酶联免疫吸附法[9]和免疫共沉淀法[10]等,但这些分析方法存在半定量或定量效率不高等问题,LC-MS/MS法与之相比,分析方法稳定、灵敏度高、选择性好、操作简便等优点[11]。
本方法首先从细胞中提取mRNA,并使用酶解法将mRNA消化为核苷,最后通过LC-MS/MS法检测样品中m6A和A的相对浓度,从而计算m6A甲基化水平。该方法直接测定核苷的相对浓度,需要核酸酶和碱性去磷酸酶将RNA水解为单个核苷,但由于酶活性和消化时间等因素可能存在RNA酶解不完全导致测定误差。为了消除这一问题,我们在预实验中考察了核酸酶和碱性去磷酸酶的反应浓度和反应时间,最终确定“1.3.1”项下RNA消化条件最为合适。为提高目标化合物的响应,对质谱参数进行了优化。ESI离子源中,在正、负离子模式下,m/z 50~400范围内进行全扫描,选取响应较强、干扰较小的作为定量离子对,以确定合适的母离子和子离子。在多反应离子监测模式下,优化了m6A,A和内标的碰撞能量,提高其响应度。最终采用m/z 282/180,m/z 268/136和m/z 152/110分别作为m6A,A和内标的定量离子对。本研究中待测物为细胞中的内源性化合物,难以获取空白基质,定量分析内源性化合物主要有代替物分析法和代替基质分析法。代替物分析法指选择合适的待测物替代分析物(如同位素等)加入真实的生物基质中配置标准曲线;代替基质分析法指待测物在替代基质(水、纯有机溶剂等)中配置标准曲线[12]。在预实验中,我们考察了同位素代替物与待测物响应值的等效性,发现两者之间有较大的差异,代替物分析法无法应用于本研究;经纯化后的待测样品基质组成较简单,主要由少量盐与蛋白酶组成,因此本研究采用了含酶和酶缓冲液的水作为代替基质测定m6A与A的浓度。在肝癌疾病中m6A是一种抑癌因子,甲基转移酶METTL-14的下调导致m6A甲基化水平降低,进而促进肿瘤细胞的转移[8,13]。采用本方法测得的肝癌细胞株HepG2的m6A甲基化水平低于正常肝细胞L02,结果与上述研究相符,提示该方法测试结果的准确性。
总之,本研究建立的LC-MS/MS法,具有快速、灵敏、准确性高、检测限低等特点,能进一步应用于m6A甲基化的生物学功能研究中。
