青少年女性四肢发作性疼痛15年
——家族性发作性疼痛综合征
2020-04-03周彬彬朱敏洪道俊
周彬彬朱敏洪道俊
1 临床资料
患者女性,16岁,因 “四肢发作性疼痛15年余”于2019年3月27日就诊于我院门诊。
患者(V:1)自8月龄始出现无明显诱因阵发性哭闹,多在下午或夜间发生,每日2~5次不等,每次约数十分钟到1 h左右,哭闹时下肢不停踢动,有时伴足部大汗。1岁半会说话时,患儿诉间断上下肢刀割样疼痛,主要位于肘、腕、膝和踝关节部位,并向肢体远端放射,每次持续30 min到1 h不等,间歇约1 h再次发作,反复3~4个周期后可自发缓解,疼痛发作1~2次每周。冬天发作时常伴随下肢出汗增多。阴雨或降温天气、疲劳等情况下易发作,尤其下雨天几乎均有发作,保暖或取暖后疼痛发作程度和频率均有减缓。疼痛发作不影响肢体运动,局部无皮疹、红肿、淤青或其他颜色改变,无关节畸形,无溃破。曾口服安乃近或布洛芬缓释胶囊有效,服药后约30 min缓解,不再发作。14岁开始,肢体疼痛频率减少,主要是下雨天或者气温明显降低时出现,但疼痛程度不减,感觉“痛时生不如死,噩梦一样疼痛”,足部出汗多,袜子湿透。患者易出现腹部痉挛性疼痛,排便或排气后好转。自幼睡眠差,入睡困难,易醒。自发病以来,生长发育基本正常,无肢体麻木,饮食尚可,无大小便功能障碍。既往史无其他特殊疾病史,无特殊药物或毒物接触史。
家族史:家系中连续五代共有15名家系成员出现类似症状(图1)。患者父亲(IV:1)42岁,自幼出现发作性肢体疼痛,20岁后疼痛症状减轻,但仍有发作,现每年发作2~3次,持续约2~3 min自发缓解,近年出现便秘症状。患者弟弟(V:2)6岁,自幼存在类似肢体疼痛症状,目前发作频繁。患者奶奶(III:2)66岁,自幼存在发作性肢体疼痛,随着年龄增长发作频率减低,28岁后完全缓解。患者奶奶的小妹(III:15)50岁,自幼存在发作性肢体疼痛,但是随年龄增长疼痛并无改变,且下肢多汗。其余10例家系成员也存在和先证者类似的肢体疼痛症状,成年患者在20~30岁之间,疼痛症状逐步消失,但有3例患者出现下肢多汗,3例患者出现便秘或肠痉挛。
体格检查:体温:36.8℃,脉搏 70次/min,呼吸:14次/min,血压 105 mmHg/70 mmHg,心律齐,叩诊心界不大,各瓣膜区听诊未闻及杂音,其他内科查体未见明显异常。发作期查体意识清楚,言语流利,高级神经活动和脑神经检查未见明确异常。四肢和躯干无明显肌肉萎缩,四肢肌力5级。双侧指鼻试验稳准,跟膝胫试验完成准确。双侧肢体感觉对称存在。腹壁反射对称引出,四肢腱反射对称引出,病理征阴性。脑膜刺激征阴性。
辅助检查:血、尿、大便常规正常,血常规、肝肾功能、血糖、血脂、电解质、肌酶谱、同型半胱氨酸、凝血功能、血沉、降钙素原、风湿抗体、C反应蛋白、抗CCP抗体、抗核抗体、铁蛋白、血清乳酸等均未见明显异常。视频脑电图监测未见癫痫样放电及慢波异常。心电图未见明显异常。肝、胆、胰、脾、双肾彩超未见异常。胸片正侧位未见异常。头颅和双膝关节MRI未见明显异常。四肢神经电图检查示运动和感觉神经传导速度、波幅、潜伏期正常。左正中神经F波出现率正常,潜伏期正常。右胫神经H反射潜伏期正常。双侧正中神经和胫神经皮肤交感未见明显异常。右股四头肌和右胫前肌针极肌电图未见异常。
病理检查:经知情同意,患者父亲自愿进行右侧腓肠神经活体组织检查。石蜡切片HE染色神经外衣和束衣结构,以及间质小血管结构正常,大有髓神经纤维未见明显异常(图2A)。Luxol快蓝染色(LFB)染色未见有髓神经纤维变性和脱髓鞘改变(图2B)。神经丝蛋白(NF)免疫组化染色显示无髓和小有髓神经轴索密度重度丢失 (图2C)。髓鞘碱性蛋白(MBP)免疫组化染色显示大有髓神经纤维密度正常,而小有髓神经纤维密度显著下降(图2D)。神经半薄切片甲苯胺蓝染色可见个别薄髓神经纤维,无髓神经纤维重度丢失(图2E)。周围神经透射电镜检查进一步证实神经束内无髓神经纤维重度丢失(图2F)。
基因检查:二代测序显示SCN11A基因上存在一个杂合突变c.665G>A,家系验证显示有症状的家系成员(V:2,V:7,IV:1,IV:6,IV:7,IV:13,III:2,III:6,III:12,III:15) 均 带有该突变,而无症状者(V:3,V:12,IV:2,IV:12,IV:16)均为野生型,提示基因型和表型呈家系共分离现象。该突变导致SCN11A蛋白第一个结构域的第IV跨膜段中高度保守的第一个精氨酸被组氨酸替代取(p.R222H),且该位点为文献已经报告的家族性发作性疼痛综合征3型(familial episodic pain syndrome-3,FEPS3)相关突变[1]。
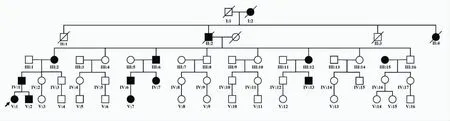
图1 家系图 箭头所示为先证者,家系内连续5代发病,呈常染色体显性遗传

图2 先证者父亲腓肠神经病理改变 HE染色神经外衣和束衣结构正常(A);LFB染色未见有髓神经纤维变性或脱鞘改变(B);NF免疫组化染色显示无髓和小有髓神经轴索密度重度丢失(C);MBP免疫组化染色显示大有髓神经纤维密度正常,而小有髓神经纤维密度显著下降(D);半薄切片甲苯胺蓝染色可见个别薄髓神经纤维,无髓神经纤维重度丢失(E);透射电镜检查显示神经束内无髓神经纤维重度丢失(F)
治疗:确诊后给予卡马西平片 200 mg,2次/d,连续2周,无效果;改用苯妥英钠片 100 mg,2次/d,连续 2周,无效果;改用草酸艾斯西酞普兰,10 mg,1次/d,气温变化时即给予对乙酰氨基酚片,0.3 g,2次/d,同时注意保暖、加强休息、心理治疗,患者疼痛症状显著缓解。2020年3月1日电话随访,患者仍在接受前述治疗方案,已有半年没有出现临床发作。
2 讨论
家族性发作性疼痛综合征是一组主要累及四肢远端,发作性的非炎性疼痛综合征,其致病机制主要和背根神经节神经元的离子通道基因变异有关[2]。按照致病基因发现的先后顺序,家族性发作性疼痛综合征分为三型:分别为TRPA1基因突变导致的FEPS1[3];SCN10A基因突变导致的FEPS2[4];以及SCN11A基因突变导致的FEPS3[1]。其中电压门控钠通道11α亚基(SCN11A)是NaV1.9钠通道核心的亚基单位,目前全世界仅有约20个FEPS3家系被报道,因而FEPS3是非常罕见的一种神经离子通道病[4-6]。我国第一批121种罕见病目录中,心脏离子通道病和先天性肌强直也是离子通道基因突变相关,然而这两类疾病患者数要远多于SCN11A突变导致的FEPS3患者。
经典的FEPS3患者通常婴幼儿期起病,表现为肢体远端的发作性疼痛。婴幼儿表述能力欠缺,因而该病的早期诊断存在一定困难[7]。然而FEPS3患儿的临床发作有一定规律可循[6]:寒冷、疲劳、饥饿、雨季是多数 FEPS3患儿的诱发因素;疼痛多在下午或夜间发生;发作性疼痛持续数十分钟后缓解,间歇一会儿后再次开始,如此反复数次,呈“串联”样痛性发作;部分患儿发作时肢体出汗增多。因此,当婴幼儿出现不明原因发作性剧烈哭闹时,如果观察到前述的一些特征,要想到是否有发作性疼痛综合征的可能形,切忌随意忽视患儿状况,甚至臆断为功能性疾病或心理方面疾病。
FEPS3患者的疼痛发作随着年龄的增长,逐步减弱并最终消失。多数患者青春期开始阶段即逐步好转,罕有超过40岁仍有发作的患者[5-7]。我们的家系患者基本上也表现为类似的疾病过程,但是有2例患者40岁后仍存在痛性发作,特别是先证者奶奶的小妹50岁时仍保持频繁的痛性发作,其可能原因与个体化的基因修饰有关,也可能与患者的环境因素有关。然而,随着病例数的积累,单基因遗传病患者表现出临床异质性是罕见病研究的一个普遍现象。此外,我们的部分家系患者出现发作时肢体出汗增多、便秘和肠痉挛等胃肠道症状,这和文献报告的相似[1,5-7]。在婴幼儿起病的发作性肢体疼痛的临床背景下,这些伴随症状可能为诊断提供有价值的线索。
患者到门诊就诊时携带了详细的风湿科、血液科、骨科相关检查,基本排外了风湿性肌痛、血小板增多症、骨髓增生性疾病、关节炎等继发性肢体疼痛疾病。该家系是我们诊断的第一个发作性疼痛综合征,鉴于有明确的常染色体显性遗传家族史,我们首先猜测的是离子通道病相关的红斑性肢痛症[8],在文献检索的过程中才知道到还有一类“家族性发作性疼痛综合征”疾病。TRPA1相关的FEPS1仅有1个家系报告,临床发作和FEPS3相似,但是疼痛部位多分布在身体上部,伴随更多的自主神经症状[3]。SCN10A相关的FEPS2也仅有1个家系报告,临床症状几乎和FEPS3一样,但是患者都在成年起病[4]。SCN11A除了与FEPS3相关之外,也与无痛症[9]、急性瘙痒症[10]、痛性周围神经病[11]、术后痛性过敏综合征[12]、小纤维神经病[13]等疾病相关。先证者父亲的周围神经病理提示重度的小纤维神经丢失,提示FEPS3患者晚期可能合并小纤维神经病。
SCN11A基因突变可以导致多种临床表型,其原因在于不同结构域氨基酸位点的变异,导致钠通道功能的丧失(loss of function)或增强(gain of function)[1]。SCN11A 蛋白由四个跨膜结构域组成,每个结构域有6段跨膜片段,其中第4段是最核心的电压感控域,其上间隔三个氨基酸即为一个正电荷氨基酸(精氨酸R或赖氨酸K),这种结构域在人类所有的钠通道α亚基都高度保守(图3)[14]。已有体外实验证明p.R222H突变,导致NaV1.9通道开放增强,细胞兴奋性增强,因而临床上表现为电兴奋增高的发作性疼痛综合征[6]。值得一提的事,我国华中科技大学的学者首先完成了SCN11A基因突变导致FEPS3的原创性发现[1]。

图3 人类不同电压门控钠通道 α亚基第一个结构域第4跨膜段是高度保守的电压感控域,其上间隔三个氨基酸即为一个正电荷氨基酸(精氨酸R或赖氨酸K),其中222位精氨酸是跨膜段的第一个正电荷氨基酸位点
离子通道病相对其他罕见病的治疗手段相对多一些。患者家系内多数患者曾接受安乃近或者布洛芬能有效减少发作次数和程度,但是不能完全阻断临床发作。患者诊断明确后,我们考虑到钠通道突变,给予作用于钠通道的抗癫痫药物,但是没有取得任何疗效,最后选择非甾体类止痛药和5-羟色胺再摄取抑制剂,取得了一定疗效。此外,在治疗SCN4A基因突变导致的周期性麻痹患者中,已经显示一定疗效的醋氮酰胺类药物是否对SCN11A基因突变有效,值得进一步观察。局部薄荷醇可阻断背根神经节中的Na v 1.8和Na v 1.9通道,其治疗作用也需要进一步研究[15]。总之,离子通道病的治疗需要结合离子通道的功能状态,结合基础研究,探索可能的治疗方案。
肢体疼痛是神经内科常见的一种临床症状,多数患者常常伴随局部的损伤或者炎症,或者明确的系统性疾病,因而相对容易确定病因。然而,当患者没有伴随这些症状和体征时,肢体疼痛的诊断有时将变得无从下手,而且部分患者存在心理异常,常常被误认为“非器质性病变”。本专栏连续报道2例以肢体疼痛为主要表现的罕见病患者,从不同的方面阐述了疼痛发生的可能原因。其实,疼痛是一种带有主观感受的防御性反应,其本质是一种电兴奋性异常的表现,当出现原发性肢体疼痛时,临床医生应该从疼痛发生和传导的解剖基础着手,重点关注外周感受器、离子通道、小纤维神经等方面的病因。
