小麦穗粒数QTL整合与元分析
2020-04-01左煜昕马靖福张沛沛栗孟飞程宏波陈思瑾杨德龙
左煜昕,马靖福,刘 媛,张沛沛,栗孟飞,程宏波,陈思瑾,幸 华,杨德龙
(1.甘肃省干旱生境作物学重点实验室,甘肃兰州 730070; 2.甘肃农业大学生命科学技术学院,甘肃兰州 730070)
穗粒数(kernel number per spike,KNS)作为小麦产量三大因素之一,是品种选育时重点考察的重要指标[1-2]。水分是影响小麦产量及稳定性的重要非生物胁迫因子,在干旱条件下,小麦穗粒数与干旱胁迫程度呈显著负相关。随着干旱胁迫的季节性频发,以及农业用水资源的日益匮乏,干旱胁迫作为主要限制因素之一,直接影响作物生长发育进程,造成穗粒数减少,最终导致减产[3]。因此,通过遗传学等方法提高穗粒数对小麦增产意义重大。
研究表明,小麦穗粒数是由微效多基因控制的复杂数量性状,遗传基础复杂,易受环境因素的影响[4]。近年来,随着DNA分子标记技术的快速发展,小麦穗粒数的分子数量遗传研究取得了较大进展。目前,研究者利用不同遗传背景材料和遗传图谱,对小麦穗粒数进行了数量性状位点(quantitative trait loci,QTL)定位和遗传分析。周淼平等[5]利用小麦重组近交系(recombinant inbred lines, RILs)群体,在1B、1D、2A、2B、3B、4A、5D、6B和7A等9条染色体上检测到11个控制穗粒数的QTL位点。吴秋红等[6]利用小麦RIL群体定位了8个控制穗粒数的QTL,位于1A、3A、3D、4A和5B等5条染色体上,可解释 4.06%~11.17%的表型变异。Lee等[7]利用小麦双单倍体群体(double haploid lines, DH)在2A、3A和4A染色体上定位了3个控制穗粒数的主效QTL。张坤普等[8]利用小麦DH群体在2D、4D和5D染色体上检测到与穗粒数紧密连锁的QTL位点。然而,由于作图群体材料遗传背景、标记类型、遗传图谱及环境条件的不同,导致小麦穗粒数定位的QTL数目、位置和遗传效应差异较大,难以获得真实、稳定的QTL位点和热点区段,无法直接应用于小麦育种实践[9]。
元分析(meta-analysis)是一种可以合并不同研究数据进行统计分析且可以对实际数据进行全面检验的方法。其中用于QTL元分析的BioMercator 4.2软件,可将与目标性状相关的所有QTL位点映射整合在一张遗传图谱上,通过比对分析,挖掘出一致性真实QTL位点,并能进一步缩小置信区间,获得与目标性状紧密连锁的分子标记。目前,该方法已广泛应用于各种作物不同性状的整合定位研究[10]。李雪华等[11]对干旱胁迫下与玉米生理性状和农艺性状相关的181个QTL整合元分析后,从中发掘出15个通用抗旱“一致性”QTL(meta quantitative trait loci, MQTL)。Goudemand等[12]利用7个DH群体对小麦叶枯病抗性相关的QTL进行定位和元分析,最终得到115个抗病QTL和27个MQTL,并发现其中14个MQTL与株高和早熟性密切相关。胡雅君等[13]通过收集涉及小麦籽粒可溶性碳水化合物含量的168个QTL,构建一致性图谱,最终获得16个MQTL。但迄今为止,有关小麦穗部相关性状的QTL整合和元分析研究相对较少,尤其是关于小麦穗粒数的QTL元分析研究未见报道。为此,本研究利用生物信息学方法,将已报道的控制小麦穗粒数的QTL位点及其数目进行收集整理,以小麦高密度遗传图谱作为参考图谱,利用BioMercator 4.2软件将QTL映射至该参考图谱上,构建小麦穗粒数QTL一致性图谱,通过元分析发掘MQTL和候选标记,为深入理解小麦穗部性状的遗传机制和精细定位提供科学依据。
1 材料与方法
1.1 材 料
以美国农业部小麦公共数据库(http://wheat.pw.usda.gov/)和已发表文献中控制小麦穗粒数的QTL信息[5-8,14-26]为研究对象。
1.2 试验方法
1.2.1 小麦穗粒数QTL数据收集整合
对已报道的控制小麦穗粒数QTL定位信息进行收集,将收集到的每个QTL数据按照BioMercator 4.2软件(http://www.mybiosoftware.com/biomercator-2-1-genetic-maps-qtl-integration.html)的要求进行整理,包括QTL名称、染色体位置、置信区间、连锁系数、贡献率、临近标记、LOD值和群体大小等,其中,QTL位置(置信区间和QTL最大可能位置)和遗传贡献率是影响QTL元分析的两个重要参数,缺一不可。
1.2.2 小麦穗粒数QTL信息映射
根据收集到的小麦穗粒数性状的QTL信息确定其所涉及的染色体,以Wheat composite 2004(https://wheat.pw.usda.gov/cgi-bin/GG3/report.cgi?class=mapdata;query=;name=Wheat,+Composite,+2004)高密度遗传图谱作为参考图谱,将目标QTL的最大可能位置和置信区间两端坐标按比例标注到参考图谱上,对比原始图谱(即所收集的遗传图谱)与参考图谱。为了获得精确的映射结果,把不能直接映射到参考图谱上的标记,可先映射到Somers等[27]绘制的小麦整合图谱(即中介图谱)上,再利用图谱上的公共标记QTL映射到参考图谱上,并将原始图谱与参考图谱上相关标记载入BioMercator 4.2软件中,建立图谱信息库。基于图谱间的共有分子标记,利用齐序函数将目标QTL的最大可能位置和置信区间两端坐标按比例标注到参考图谱上,即映射,并将原始图谱及参考图谱间有争议的标记剔除。
1.2.3 小麦穗粒数QTL元分析
利用BioMercator 4.2软件对小麦穗粒数QTL进行元分析,将位于同一连锁群相同位点附近的N个独立存在的与目标性状相关的QTL进行运算,对独立来源的同一性状且位于同一座位或有重叠座位的QTL计算出一个MQTL,该QTL会给出5个模型(即模型1、2、3、4和N),其中赤池信息量准则(akaike-type criteria values, AIC)值最小的模型为最优模型,即真实QTL模型,并通过高斯定理最大似然比估算该QTL存在的位置和置信区间。如果AIC值最小的为N模型,则表明用于分析的连锁群过大,需要分为两段来进行元分析。如果整理数据时某一QTL置信区间未知,可通过Darvasi等[28]应用的公式推断95%的置信区间:
C.I=530/(N×R2)
(1)
C.I=163/(N×R2)
(2)
其中C.I指QTL 95%的置信区间,N代表作图群体的大小,R2代表该QTL的遗传贡献率,公式(1)适用于F2群体和BC群体,公式(2)适用于RIL、DH及NIL群体。若已知置信区间,也可应用该公式估算未知QTL的遗传贡献率。
1.2.4 基于小麦穗粒数MQTL范围内候选基因发掘
利用生物信息学手段进行目标性状QTL的整合,获得穗粒数MQTL区域,针对这些区域内的EST或DNA序列搜索候选基因。该法的原理为:物种内及物种间的序列同源性,基因的功能和序列是密切相关的,当序列的相似性超过一定的范围时,它们可能执行相同的功能,通过将未知功能序列和已知功能序列的对比,如果它们相似性较高,就可以推断出序列的相应功能。
在参考图谱Wheat composite 2004上,由元分析所得的小麦穗粒数MQTL区域(meta-C.I)由两端标记界定。对MQTL区间及其邻近区域的穗粒数相关基因位点进行整理,根据MQTL区间的基因位点名称,在GrainGenes(https://wheat.pw.usda.gov/)网站,下载目标性状“一致性”区段内的相关基因序列和各种标记的原始序列,从而确定MQTL在染色体物理图谱上的位置。利用小麦基因组数据库(http://202.194.139.32/)中JBrowse工具检索MQTL内的基因信息,并获得该区间内所有基因的功能注释信息。最后在NCBI网站上下载这些基因的序列,并利用其在线工具BLAST分析比对相关基因序列,进而预测目标性状候选基因。
2 结果与分析
2.1 小麦穗粒数QTL信息的收集整合结果
从小麦公共数据库和已发表文献中收集到来源于花培3×豫麦57、Keumkang×Olgeuru、小麦-冰草衍生系3228×京4839、川麦42×川农16、洛旱2×潍麦8、潍麦8×烟农19、潍麦8×济麦20、望水白×Alondra、兰考906×小偃21、G1816×Langdon、西农817×中国春、燕达 1817×北农6、扬麦17×宁麦18、糯麦1×藁城8901、TP×Ta13等18个作图群体,共涉及小麦穗粒数的163个QTL(表1)。利用BioMercator 4.2软件中的QTL projection功能将收集到的数据映射至Wheat composite 2004小麦参考图谱上。LOD值在2.01~41.13之间,各位点的遗传贡献率在2.20%~32.75%之间。
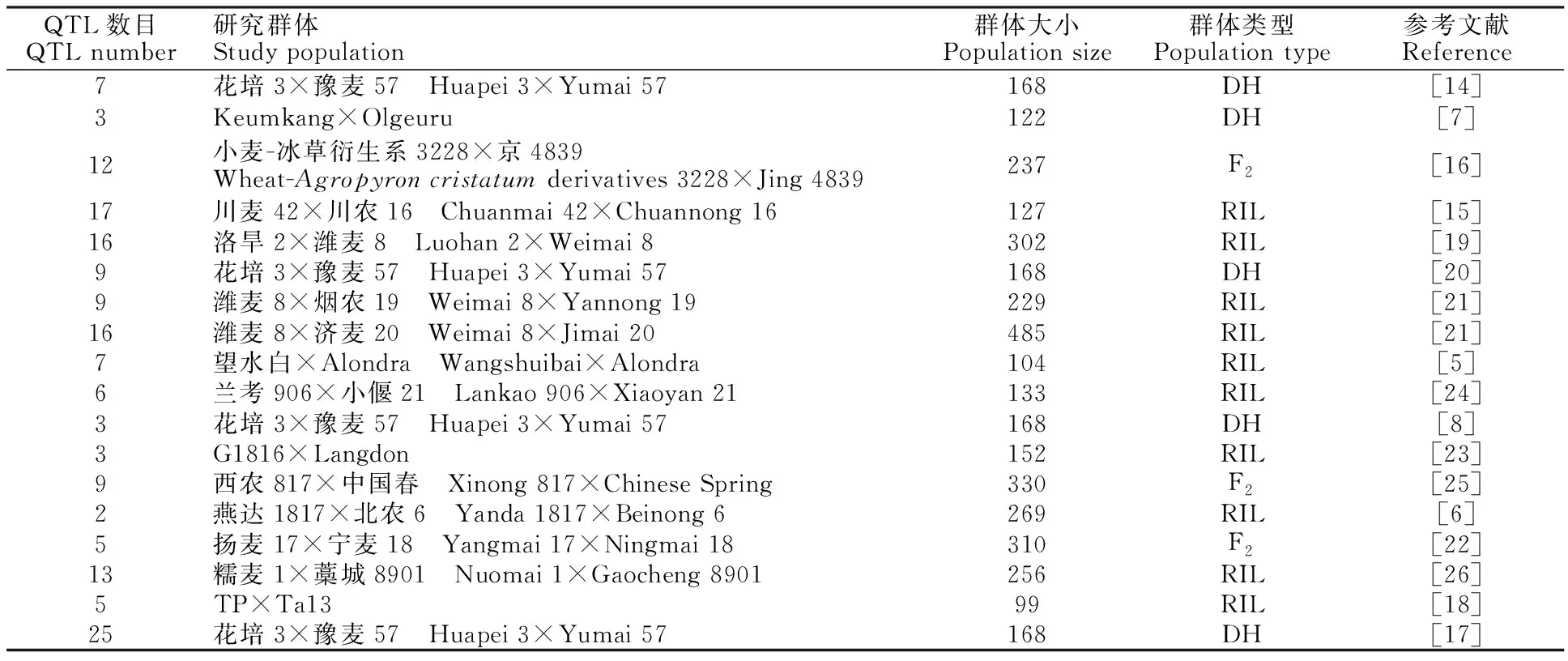
表1 小麦穗粒数QTL数据整合Table 1 Integration of QTL data for the kernel number per spike in wheat
2.2 小麦穗粒数QTL一致性图谱的构建
将原始图谱与参考图谱Wheat composite 2004 上相关标记载入BioMercator 4.2软件,利用图谱映射程序构建小麦穗粒数QTL一致性图谱。结果(图1)表明,控制小麦穗粒数的QTL覆盖了小麦21条染色体,各染色体上分布不均,在7D染色体上最少(2个QTL),2B染色体上最多(17个QTL),其他染色体上分布3~12个。其中,在QTL一致性图谱的同一连锁群上有明显成簇分布现象,存在QTL富集区域。如在2A、3A和7B连锁群上各有2个QTL簇,在2D、3D和6B连锁群上各有1个QTL簇。这些QTL彼此间有区间的重叠,大都出现在一段区间内,如在3A连锁群的80 cM左右有8个QTL,说明该区间很可能是控制目标性状的热点区域并存在更为真实重要的QTL,含有大量控制小麦穗粒数的基因(图1)。

染色体左侧“点”至“横线”表示QTL所在位点的遗传贡献率大小的连续变化;“竖线”表示QTL所在置信区间。The “dot” to “transverse line” on the left side of chromosome means the successive change in genetic contribution rate of QTL; “Vertical line” means the confidence interval of QTL.图1 小麦穗粒数QTL一致性图谱Fig.1 Consensus map of QTL for kernel number per spike in wheat
2.3 小麦穗粒数QTL元分析
结合小麦已定位的QTL信息,利用BioMercator 4.2软件中元分析程序分析各连锁群上的QTL。由于分析模型不同,以每次分析中AIC值最小的区间为最优,确定1个真实QTL,最终共得到35个控制小麦穗粒数的MQTL(表2),分别位于小麦的1D(2个)、2A(4个)、2D(4个)、3A(4个)、3D(3个)、4D(4个)、5A(4个)、6B(4个)、7B(4个)和7D(2个)染色体上,平均每条染色体上含有3.4个MQTL。
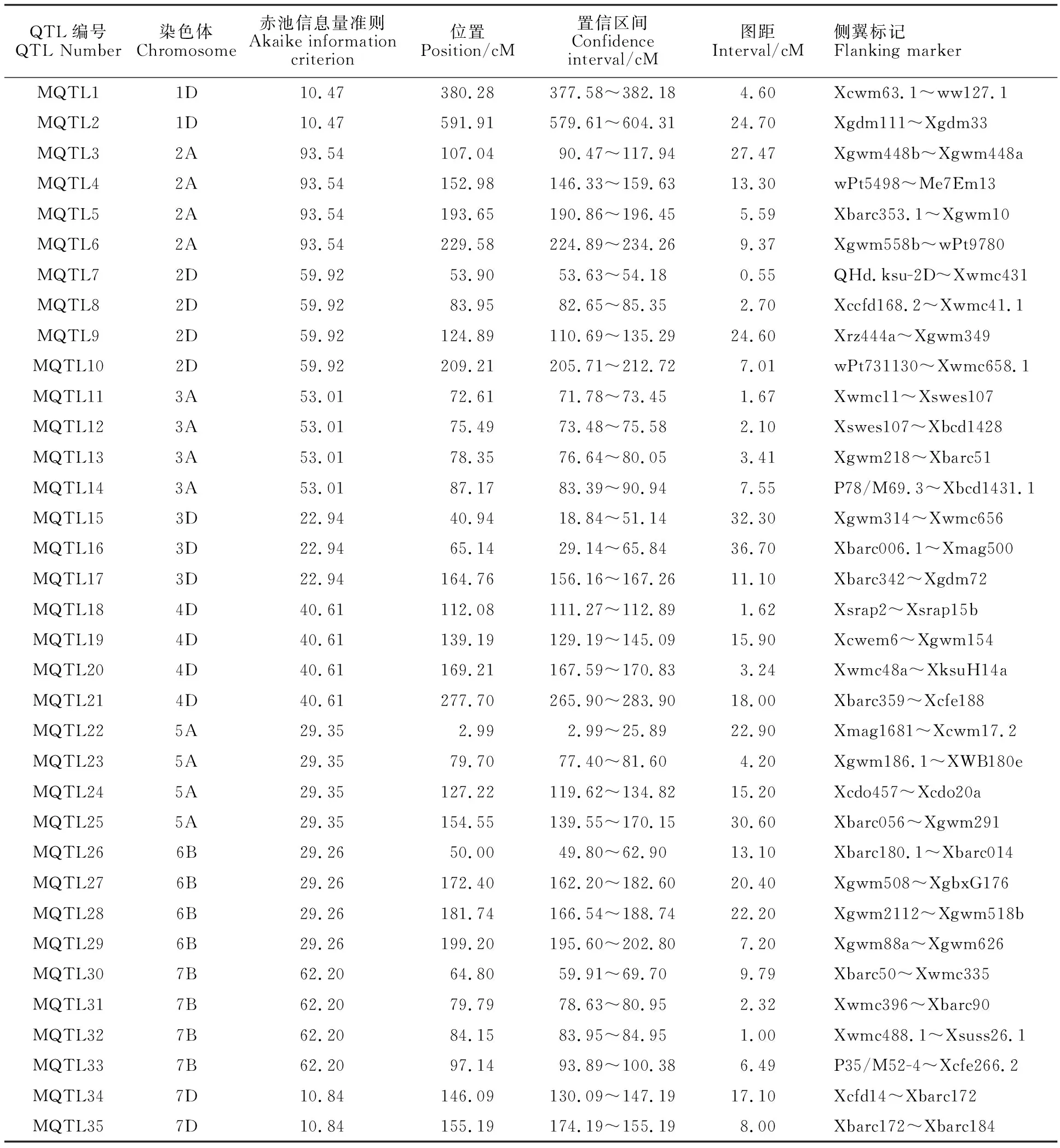
表2 小麦穗粒数QTL的元分析Table 2 Meta-analysis of QTL for the kernel number per spike in wheat
将35个控制小麦穗粒数的MQTL按照其所在染色体的位置依次排序为MQTL1~MQTL35,其中有7个MQTL的置信区间小于 3 cM,分别是MQTL7(0.55 cM)、MQTL8(2.70 cM)、MQTL11(1.67 cM)、MQTL12(2.10 cM)、MQTL18(1.62 cM)、MQTL31(2.32 cM)和MQTL32(1.00 cM)。经元分析后,这些检测到的MQTL所存在的位置和置信区间均优化了原QTL的位置和效应,缩小了原置信区间,检测出更为精确的MQTL,很大程度上减小了由于不同试验所得到的QTL位置差异而造成的误差,提高了QTL定位的准确度和有效性。其中,4个MQTL均分布在3A染色体的相邻区域,并且其图距均小于10 cM (1.67~7.55 cM);除此之外,MQTL27(162.20~182.60 cM)与MQTL28 (166.54~188.74 cM)的置信区间有很大的重合,这表明这些区段很可能对小麦穗粒数具有重要贡献。
2.4 小麦穗粒数MQTL范围内候选基因预测
根据QTL一致性区间内的SSR标记所在参考基因组中的位置,利用小麦基因组数据库所提供的基因注释结果和基因预测,对小麦穗粒数定位区域进行了候选基因的预测。由于较小的QTL置信区间有利于提高QTL定位的准确度和有效性,根据“一致性”QTL区间两端标记在小麦物理图谱中的位置,将小麦穗粒数定位结果中图距较小较精确的MQTL位点进行物理图谱定位,统计MQTL区间内所包含的基因个数。结果发现,在2D染色体上,与小麦穗粒数性状紧密连锁的两个分子标记在Xcfd168.2~Xwmc41.1之间(即MQTL8),其遗传距离为82.65~85.35 cM(≈2.57 Mb)范围内,共包含112个基因,单位长度内基因个数为43.6个。
由表3可以看出,本研究共发现了4个与目标性状相关的候选基因,这些基因座位涉及信号传导、渗透调节和糖代谢等多种生理生化途径。YUC基因家族编码类黄素单氧化酶,是IAA生物合成途径中催化色胺的N-氧化反应的限速酶,对生长素的合成起重要调控作用;SUS蔗糖合酶基因作为籽粒中糖积累转运的重要代谢基因,对籽粒产量有重要影响;ERF家族转录因子是植物中重要的一类转录因子,广泛参与植物各类生理过程。对目标性状MQTL8的置信区间进行候选基因筛选,获得CKX基因家族成员。研究表明,CKX作为降解细胞分裂素(cytokinins)的一种黄素酶,广泛分布于植物各个部位,CKX对植物配子发育和作物产量的形成具有显著的影响。以上候选基因均为与小麦产量可能相关的基因,在一定程度上对改善产量发挥重要作用,然而具体功能有待进一步的试验验证。

表3 小麦穗粒数MQTL内相关候选基因信息Table 3 Candidate genes within MQTLs related to the kernel number per spike in wheat
3 讨 论
3.1 图谱的选择
元分析中最重要的就是分子标记遗传参考图谱的选择。在小麦相关性状元分析的过程中最常用的是以11张标准图谱整合的Wheat composite 2004,该图谱共涉及3 741个标记,所包含的分子标记主要是以简单重复序列(SSR)、扩增片段长度多态性(AFLP)和限制性内切酶片段长度多态性(RFLP)组合构建,总长3 236 cM。该图谱与已完成和正在绘制的QTL定位图谱间存在较多共同标记,但是研究发现,某些研究中原始图谱的标记仍与参考图谱中标记一致性较差,个别目标性状QTL不能直接映射到该参考图谱上,因此,本研究使用Somers等[27]绘制的小麦整合图谱作为中介图谱,该图谱与Wheat composite 2004参考图谱存在许多共同标记,并且标记覆盖面较广,提高了目标性状图谱与参考图谱间的映射,缩短了两标记之间的距离,为精细定位提供良好的基础,保证了QTL的整合分析。
3.2 元分析应用
近年来,利用元分析方法对玉米、大豆等作物相关性状QTL的遗传改良取得了一定进展。方永丰等[29]整合了173个与玉米持绿性相关的QTL,发掘出5个持绿MQTL区间,并在MQTL区域内发掘出8个持绿相关候选基因;王晓丽等[30]构建了含221个玉米产量及构成因子QTL的整合图谱,并在玉米6号染色体上确定了一个与穗数、粒重和单位籽粒产量均相关的MQTL。吴 琼等[31]通过对来自10个不同群体的与大豆生育期有关的98个QTL进行元分析,最终获得了9个MQTL及其连锁标记。当大量控制目标性状的基因或QTL被整合到一致性图谱上后,控制相同和不同的目标性状的QTL分布特征更加明显。但是利用元分析手段对小麦数量性状进行QTL整合研究的报道较少,尤其是小麦穗粒数的元分析研究还相对滞后。随着小麦全基因组信息的完善以及物理图谱的成功构建,对于QTL热点区域和被前人反复证实的小麦目标性状QTL的核心染色体区段,将是发掘小麦关键基因的重点研究部位,可为小麦克隆及分子改良育种提供大量侯选基因。
本研究整合了近年发表的163个控制小麦穗粒数的QTL,构建了小麦穗粒数QTL一致性图谱,具有簇集分布的特征,并通过元分析,得到了35个MQTL及其紧密连锁标记,其置信区间最小可缩小到0.55 cM,明显减小了QTL的误差,使结果更准确可靠。其中,本研究在3A染色体上P78/M69.3~Xbcd1431.1区间内获得的MQTL与叶亚琼等[32]在该区域发现的控制株高的MQTL享有相同的标记区间。Tuberosa等[33]认为,这种现象可能与“一因多效”或者控制不同性状的基因紧密连锁有关,这可为图谱构建以及寻找和定位同源基因提供切入点。本研究所发掘的MQTL是由不同作图群体材料整合的,后续可以利用分子标记辅助选择技术(marker assisted selection, MAS),对后代进行基因型预测,进而提高MAS的实际效率。
3.3 相关基因发掘
随着小麦全基因组测序的完成,各种生物信息的迅速增长,对未能精细定位的基因提供了越来越多的研究策略和思路。在MQTL区域内可以获得许多DNA序列,这些序列具有较完整的基因结构,从而可以对相关基因进行预测。同时也可以通过对蛋白序列的同源比对,以此来确定基因的功能[34]。
通过在QTL置信区间进行候选基因预测,可以在一定程度上验证QTL定位的准确性。在本研究中,充分利用不同的研究成果以及公共信息资源,对不同时期、不同环境条件和不同研究群体下定位的小麦穗粒数QTL进行整合优化,得到了MQTL。利用两端标记在物理图谱中的位置,初步预测MQTL区域内的相关基因。最终,在1个MQTL区间(MQTL8)内初步确定4个与产量性状相关的候选基因,这些基因座位涉及信号传导、渗透调节和糖代谢等多种生理生化途径。其中,通过对候选基因序列同源比对,发现TraesCS2D01G587100LC.1基因与TaCKX基因[35]的序列同源性较高(91%),因此我们推断该基因也与小麦籽粒发育可能紧密相关。研究表明,小麦CKX是由一系列同工酶组成的[35],具有调控小麦不同时期生长发育的功能,TaCKX与水稻、玉米和大麦等作物CKX基因具有较高的同源性。目前已知TaCKX基因与许多产量性状关系密切,最新研究表明该基因的等位变异与叶绿素含量也紧密相关[36]。因此,在后续研究中,可以对该基因进行本体分析、转录调控分析以及代谢途径分析,进一步为精细定位和图位克隆奠定基础。试验证明,利用元分析手段可获得较小置信区间的MQTL,然后映射到物理图谱上,可以将目的基因锁定在一个较小的范围内。再针对目标区段构建回交群体,根据基因的保守性进行目标基因的发掘,通过这种方法可以提高基因克隆的效率。
