先天性挛缩蜘蛛指畸形两个家系报道并文献复习
2020-03-29殷耀斌侯春梅田文赵俊会
殷耀斌 侯春梅 田文 赵俊会
先天性挛缩蜘蛛指畸形 ( congenital contractural arachnodactyly,CCA ) 或者 Beals Hecht 综合征 ( Beals Hecht syndrome,BHS ),是一种罕见的常染色体显性遗传的结缔组织疾病。自 1971 年 Beals 和 Hecht 报道此畸形以来,国内外关于此种畸形均为小样本病例或者个案报道,我院收治 2 个 CCA 家系病例,结合文献复习报告如下。
临床资料
家系 1:
患儿,女,4 岁 8 个月,出生时便发现患儿双手、肩关节、肘关节及膝关节屈曲挛缩,外耳呈现异常褶皱,四肢细长,脊柱侧凸,有鸡胸畸形。患儿母亲为改善患儿手部挛缩而来我院治疗。入院后查体:患儿身高 1.1 米,体重 16 kg,患儿体型细长,肩关节最大外展只能到 90°,肘关节伸直差 30°,手指细长,屈曲挛缩,以近指间关节最重,伸直差 70°。膝关节屈曲挛缩,伸直差 20°;足趾屈曲挛缩,以第二足趾最重。外耳廓呈现异常褶皱。头颅未见明显异常。胸部呈鸡胸畸形,脊柱侧凸畸形。入院行心脏彩超未发现心脏畸形。患儿母亲产前检查时便发现胎儿异常,患儿孕 37 周经剖腹产出生,出生体重 3.6 kg,身高 50 cm。患儿母亲叙述患儿语言能力及智力发育水平低于同龄孩子 ( 图1 )。
患儿家族情况。该家族最初患病者为此患者曾外祖父,其曾外祖父育有五女一子,其中有二女为CCA 患者,其中之一便是患儿外祖母。患儿外祖母生育四女,其中有二女为 CCA 患者,其中之一便是患儿母亲。患儿母亲育有三女一儿,其中一儿一女为 CCA 患者。而患儿母亲的妹妹,育有一儿一女,其中女儿为 CCA 患者。家族遗传图 ( 图2 )。该家系除了手部畸形外,耳部特有畸形最为明显 ( 图3 )。
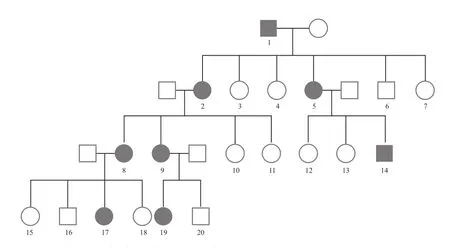
图2 家系 1 家族遗传情况,序号 17 为患儿Fig.2 Genetic map of the first family. The patient was Number 17
家系 2
患儿,男,7 岁,出生时便出现双手关节屈曲挛缩畸形,随着生长发育挛缩畸形加重来我院就诊。患儿身高 1.3 米,体重 30 kg,患儿体型细长,肩关节最大外展只能到 100°,肘关节伸直差 10°,手指细长,屈曲挛缩,以近指间关节最重,伸直差 70°。膝关节屈曲挛缩,伸直差 15°;足趾屈曲挛缩不明显。外耳廓呈现异常褶皱。头颅未见明显异常。入院行心脏彩超未发现心脏畸形。患儿母亲叙述患儿语言能力及智力发育水平低于同龄孩子 ( 图4 )。
家族情况:患儿家族的始发病者是患儿父亲。患儿父亲也合并有严重的手部屈曲挛缩畸形,接受了手术治疗。患儿父亲有一子一女,均为 CCA 患者( 图5 )。

图3 a:为图2 中家族序列 2 的耳部畸形;b:为图2 中家族序列 8 的耳部畸形;c:为图2 中家族序列 9 的耳部畸形;d:为图2 中家族序列 14 的耳部畸形Fig.3 a: Auricle malformaition of fig 2, No. 2; b: Auricle malformaition of fig 2, No. 8; c: Auricle malformaition of fig 2, No. 9; d: Auricle malformaition of fig 2, No. 14
文献检索
以“先天性挛缩蜘蛛指畸形”、“Congenital contractural arachnodactyly”、“Beals Hecht syndrome”为关键词在中国知网,万方数据库,维普数据库,PubMed 数据库进行检索,去除重复文章,共搜索到 142 篇文章,大多数为个案报道,共有 33 个 CCA 的家系报道,其中有 6 个中国家系的报道。

图4 a:患儿身材纤细;b:患儿耳部畸形;c:患儿手部屈曲挛缩畸形;d:患儿父亲耳部畸形;e:患儿父亲手部畸形Fig.4 a: The patient was slender; b: Auricle malformaition; c: Arachnodactyly and joint contracture; d: Auricle malformaition of the patient’s father; e Hand deformity of the patient’s father

图5 家系 2 始发病者为患儿父亲,患儿有一姐姐,同样是 CCA患者Fig.5 The proband of the second family was the father of the patient.The patient had an older sister, who was also a CCA patient
讨 论
一、病因学
CCA 最早由 Beals 和 Hecht 于 1971 年报道并命名。此综合征临床表现与马凡综合征高度相似,所以马凡综合征的发现者 Antoine Marfan 所报道的病例其实是 CCA[1]。马凡综合征的致病基因是原纤蛋白 1基因 ( FBN1 gene ),是 1991 年 Dietz 运用原位杂交技术定位并克隆的。而 Beals Hecht 综合征是由常染色体 5q23 上的原纤蛋白 2 基因 ( FBN2 gene ) 突变导致此综合征的发生[2-8]。FBN2 基因是在研究马凡综合征基因病变基础时被发现的。原纤蛋白是一种富含半胱氨酸的糖蛋白,它是微纤维的重要成分,在纤维原形成过程中起到至关重要的作用。原纤蛋白由纤维母细胞分泌至细胞外基质,整合于构成弹性蛋白沉积支架的微纤维上。FBN1 基因所产生的蛋白在结构、数量和序列上均与 FBN2 相类似。然而,FBN1 与 FBN2 在功能上均不相同,在不同的发展阶段与组织分布中,FBN1 与 FBN2 的表达各有其特点。原纤蛋白 2 在胚胎发育过程中弹性纤维的聚集发挥作用,而原纤蛋白 1 构成了支撑组织器官的框架结构。原纤蛋白 2 基因的突变导致原纤蛋白 2中心区域结构发生改变。在 CCA 患者中仅有一部分 ( 25%~75% ) 可发现原纤蛋白 2 基因突变[9-10]。马凡综合征基因突变遍布整条 FBN1 基因链,目前报道的突变类型超过 1000 种;而 CCA 基因突变主要集中在 FBN2 基因中间区域,突变类型大约 40 余种[9,11]。在 CCA 家族中父母存在体细胞和生殖细胞嵌合现象,这也是 CCA 家族中不同患病个体表现不一的原因。本篇报道了 2 个家系由于患者拒绝行基因检测,所以无法从基因学上进行确诊,是本报道的不足之处。
二、临床分型及特点
临床上将 CCA 分为经典型和严重性 / 致死型两种类型。对于经典型 CCA,主要包括骨骼畸形、面部畸形及心脏畸形。
1. 骨骼畸形:( 1 ) Marfan 综合征样外形,例如:瘦长体型,臂展长度大于身高 ( 但这点在 CCA 患者中由于关节挛缩的原因,表现并不明显 ),手指及足趾细长[12];( 2 ) 肩、肘、腕、髋、膝关节及近指( 趾 ) 间关节挛缩畸形,有的患者有扣拇畸形和 ( 或 )足内翻畸形。随着生长发育,大关节的挛缩会有所缓解,但手部挛缩会持续存在;( 3 ) 四肢肌肉发育不良;( 4 ) 脊柱后突或者侧凸畸形,出生时便可出现,而且随着生长发育可能会逐步加重[13];Meena 等[14]报道了 1 例 CCA 患者出现枕骨大孔狭窄合并 C2和 C3融合的病例;( 5 ) 有的患者合并鸡胸或者漏斗胸。由于胸部畸形及脊柱畸形,引发肺部通气障碍,因而有反复肺部感染、呼吸窘迫的报道[15],对于 CCA 患者进行全身麻醉时要格外注意呼吸道的监管。
2. 面部畸形:( 1 ) 外耳廓的特有畸形,表现为外耳廓上部褶皱畸形,这一表现往往是 CCA 所特有的,可作为鉴别诊断的重要依据;本研究中报道了典型的外耳畸形,期望加深临床医生对 CCA 的印象;( 2 ) 高额弓畸形、舟状头畸形、长头畸形、短头畸形、额隆起畸形等,这些畸形虽不常见,但在 CCA 病例中确有报道;( 3 ) 眼部畸形,在 CCA 患者中仅有 20% 出现眼部畸形,例如:蓝色巩膜,轴性近视,白内障,晶状体裂,睫状体发育不良,青光眼[16]。
3. 心脏畸形:通常表现为室间隔缺损,主动脉发育异常和单脐动脉等,而主动脉根部非进行性扩张和Valsava 窦扩张并不常见。到目前为止,仅有 1 例 CCA患者出现主动脉扩张合并主动脉夹层的报道[17]。
对于严重 / 致死型 CCA,除了具备经典型 CCA的临床表现外,往往合并有严重的心脏畸形或者胃肠道畸形。心脏畸形包括:二尖瓣脱垂,主动脉基底扩张,房间隔缺损,室间隔缺损。胃肠道畸形包括十二指肠闭锁、食道闭锁、肠扭转等。虽然临床病例有限,但这类患者往往在出生后 1 周内进行手术治疗,呼吸道并发症是临床致死的主要原因。
除了常见的临床表现外,有 CCA 患者存在智力发育延迟和自闭症的个案报道[18-19]。Callewaert 报道的 32 例 CCA 患者中,有 2 例有股四头肌发育不良合并髌骨松弛,有 2 例皮肤菲薄,圆锥角膜和肾盂输尿管交界区域狭窄。
三、鉴别诊断
临床上,CCA 与马凡综合征有着许多相似的临床特点,但仍存在一些鉴别要点:( 1 ) 与马凡综合征相比,CCA 患者心脏畸形很少出现主动脉基底部明显扩张[20];( 2 ) 马凡综合征患者出现晶状体移位的几率远高于 CCA[21];( 3 ) CCA 患者外耳特有的褶皱是其区别与其它疾病的重要鉴别点。另外 CCA 尚需和一些罕见的综合征进行鉴别,如 Loeys-Dietz 综合征[22]。
四、治疗和产前监控
CCA 患者畸形表现各异,需要根据患者具体情况设计个性化的治疗方案。由于患者四肢大关节屈曲挛缩随着生长发育而逐步缓解,往往无须针对性治疗。而手部关节挛缩往往持续存在并会影响手部外观及功能,故需要进行关节松解及取皮植皮来改善功能及外观。由于脊柱后突和侧凸畸形并不能随着生长发育而缓解,故可能需要手术矫正。而心脏畸形严重者往往需要早期手术治疗,对于不严重者需要定期随访监测。
由于 CCA 是一种常染色体显性遗传疾病,患者下一代发生率为患者的概率是 50%,因而对于有CCA 家族病史的孕妇进行产前检查至关重要。Inbar-Feigenberg 等[23]报道了 2 例 CCA 胎儿产前 B 超影像表现为手指始终呈握拳位而且多发关节屈曲位,且这一表现在不同孕期检查时一直持续存在。
总之,CCA 是一种罕见的常染色体显性遗传的结缔组织疾病,诊断主要依靠临床表现及基因诊断。
