补肝益肾颗粒质量标准研究
2020-03-20张靖年毕晓黎胥爱丽李养学李素梅蓝森梅
张靖年毕晓黎胥爱丽李养学李素梅蓝森梅
(1.广东省中医药工程技术研究院/广东省中医药研究开发重点实验室,广东 广州510095;2.中山大学新华学院,广东 广州510520)
补肝益肾颗粒原方系广东省第二中医院骨科专家常用的临床验方,由淫羊藿、山药、续断、白芍、泽泻、制何首乌等8 味中药组成,方中淫羊藿补肝肾之阳[1],为君药。 温性之杜仲[2]、续断[3]、桑寄生[4]为臣药,能补肝肾、强筋骨。 佐以制何首乌补益精血[5],泽泻利湿而泄肾浊[6],白芍柔肝敛阴[7]。 全方肝肾同补,补中寓泄,共奏补益肝肾作用。 该方临床疗效良好,但汤剂存在煎煮、携带、使用不方便的特点。 现将其制成颗粒剂,既保持原汤剂的特色和优势,又具有体积小、利于储藏、便于携带、使用方便等优点。 但该制剂目前尚无质量标准,为较好地控制其质量以保证临床疗效,本试验采用TLC 法定性鉴别方中君药淫羊藿、佐药白芍、制何首乌等药味。研究表明,淫羊藿苷具有改善骨代谢、治疗骨质疏松症等药理作用[8-9],还可改善生殖功能[10-11]、保护肾脏[12];川续断皂苷Ⅵ对成骨分化具有促进作用[13-14],并具有一定的肝脏保护作用[15-16],这与补肝益肾颗粒的功效有一定的关联性。 因此,本试验采用HPLC 法定量测定君药淫羊藿所含淫羊藿苷、臣药续断所含川续断皂苷Ⅵ的质量分数,以期为本品质量控制提供实验依据。
1 仪器与试药
1.1 仪器
CAMAG TLC SAMPLER 4 型自动点样仪,CAMAG REPROSTAR 3 型薄层成像系统(瑞士CAMAG 公司);Waters e2695 Sepatation Module 高效液相色谱仪,Waters 2998 PDA 检测器,四元梯度泵,Empower Pro 色谱工作站(美国Waters 公司);KQ5200DE 型超声波清洗器(昆山市超声仪器有限公司);METTLER XS205DU 型电子分析天平(瑞士Mettler-Toledo 公司);HH-8 数显恒温水浴锅(上海梅香仪器有限公司);MilliPore Advantage A10 自动纯水机(美国MilliPore 公司)。
1.2 试药
乙腈等HPLC 所用试剂均为色谱纯(德国默克公司);水为屈臣氏蒸馏水;其他试剂均为分析纯(广州化学试剂厂)。
补肝益肾颗粒(批号:20181004、20181005、20181006)由广东省第二中医院制剂室提供;淫羊藿对照药材(批号:121632-201502)、白芍对照药材(批号:120905-201610)、制何首乌对照药材(批号:121454-201405)、淫羊藿苷对照品(批号:110737-201516,质量分数94.2%)购自中国食品药品检定研究院;川续断皂苷Ⅵ对照品(批号:C-014-170717,质量分数98.3%)购自成都瑞芬思生物科技有限公司。
2 方法与结果
2.1 定性鉴别
2.1.1 淫羊藿的鉴别取本品2 g,加水20 mL 使溶解,用乙酸乙酯振摇提取2 次,每次20 mL,混合乙酸乙酯液,蒸干,残渣加乙醇1 mL 使溶解,作为供试品溶液。 另取淫羊藿对照药材0.4 g,加水适量,加热至沸腾并保持微沸30 min,滤过,滤液浓缩至约20 mL,自“用乙酸乙酯振摇提取2 次”起,同法制成对照药材溶液。 再取除淫羊藿外处方比例的其余7味中药,按处方工艺制备阴性样品,取阴性样品2 g,自“加水20 mL 使溶解”起,同法制成淫羊藿阴性对照溶液。 按照薄层色谱法[17]试验,吸取上述溶液各3 μL,分别点于同一硅胶H 薄层板上,以乙酸丁酯-甲酸-水(体积比13 ∶10 ∶10)的上层溶液为展开剂,展开,取出,晾干,喷以AlCl3试液,105 ℃加热2 min,置紫外光灯(365 nm)下检视。 供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,阴性对照无干扰。 见图1。
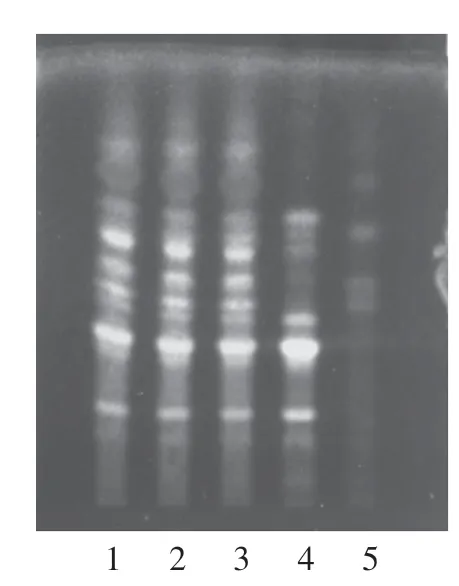
图1 淫羊藿鉴别的薄层色谱图Figure 1 TLC of Epimedii Folium
2.1.2 白芍的鉴别取本品4 g,研细,加乙醇25 mL,超声处理30 min,滤过,滤液蒸干,残渣加水20 mL使溶解,用水饱和正丁醇溶液萃取2 次,每次20 mL,混合水饱和正丁醇液,蒸干,残渣加乙醇1 mL 使溶解,作为供试品溶液。 另取白芍对照药材1 g,加水适量,加热至沸腾并保持微沸30 min,滤过,滤液浓缩至约20 mL,自“用水饱和正丁醇溶液萃取2 次”起,同法制成对照药材溶液。 再取除白芍外处方比例的其余7 味中药,按处方工艺制备阴性样品,取阴性样品4 g,自“加乙醇25 mL”起,同法制成白芍阴性对照溶液。 按照薄层色谱法[17]试验,吸取供试品、对照药材及阴性对照3 种溶液各5 μL,分别点于同一硅胶G 薄层板上,以三氯甲烷-乙酸乙酯-甲醇-甲酸(体积比40 ∶5 ∶10 ∶0.2)为展开剂,展开,取出,晾干。喷以5%(质量浓度)香草醛硫酸溶液,105 ℃加热约1 min。 供试品色谱中,在与对照药材色谱相应的位置上,显相同的蓝紫色斑点,阴性对照无干扰。 见图2。
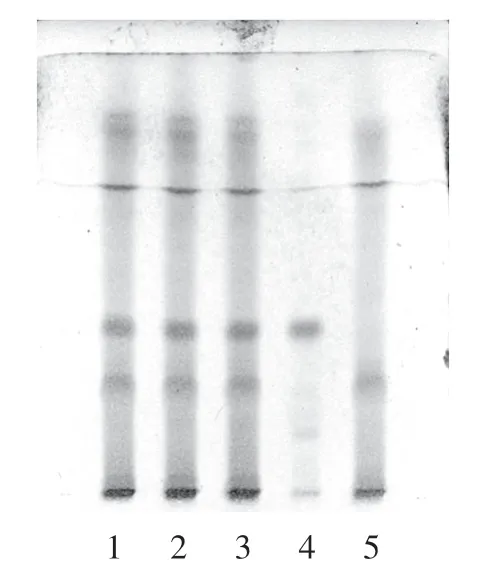
图2 白芍鉴别的薄层色谱图Figure 2 TLC of Paeoniae Radix Alba
2.1.3 制何首乌的鉴别取本品4 g,加水30 mL 使溶解,用乙醚振摇提取2 次,每次30 mL,混合乙醚液,蒸干,残渣加甲醇1 mL 使溶解,作为供试品溶液。 另取制何首乌对照药材1 g,加适量水煮沸30 min,滤过,滤液浓缩至约30 mL,自“用乙醚振摇提取2 次”起,同法制成对照药材溶液。 再取除制何首乌外处方比例的其余7 味中药,按处方工艺制备阴性样品,取阴性样品4 g,自“加水30 mL 使溶解”起,同法制成制何首乌阴性对照溶液。 按照薄层色谱法[17]试验,吸取上述溶液各10 μL,分别点于同一硅胶G 薄层板上,以环己烷-二氯甲烷-乙酸乙酯-甲酸(体积比10 ∶1 ∶1 ∶0.1)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。 供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点,且阴性对照无干扰。 见图3。
2.2 定量测定
2.2.1 对照品溶液的制备取淫羊藿苷、川续断皂苷Ⅵ对照品适量,精密称定,加甲醇制成每1 mL 分别含淫羊藿苷、川续断皂苷Ⅵ254.75、113.75 μg 的混合对照品溶液,即得。

图3 制何首乌鉴别的薄层色谱图Figure 3 TLC of Polygoni Multiflori Radix Praeparata
2.2.2 供试品溶液的制备取本品适量,研细,精密称取1 g,加甲醇25 mL,称定质量,水浴加热回流处理60 min,放冷,再称定质量,用甲醇补足减失的质量,摇匀,滤过,取续滤液,即得。
2.2.3 空白溶液的制备取甲醇溶液适量,滤过,取续滤液,即得。
2.2.4 色谱条件与系统适用性研究色谱柱:phenomenex Luna Omega Polar C18(4.6 mm×250 mm,5 μm)柱;流动相:乙腈(A)-水(B):0 ~20 min,25%A→30%A;20 ~30 min,30%A;柱温:25 ℃;流速:1.0 mL/min;检测波长:212 nm;进样量:10 μL。分别精密吸取对照品、供试品、空白溶液10 μL,按上述条件测定,理论塔板数按淫羊藿苷计,不低于3 000;分离度>1.5。 见图4。
2.2.5 线性关系考察分别精密称取淫羊藿苷、川续断皂苷Ⅵ10.19、4.55 mg,置于同一10 mL 容量瓶中,加甲醇溶解并稀释定容至刻度,摇匀,记编号MS1。 精密吸取MS1 5 mL 置于10 mL 容量瓶中,用甲醇稀释定容至刻度,记编号MS2;依次倍比稀释制得MS3~MS6。 分别精密吸取上述6 种混合对照品溶液各10 μL,注入高效液相色谱仪,按“2.2.4”项下色谱条件测定。 以对照品进样量X为横坐标,对应的峰面积Y为纵坐标,绘制标准曲线并进行线性回归,得到淫羊藿苷、川续断皂苷Ⅵ的回归方程分别为Y1=1.998 5×105X1-7.369 4×104(r1=0.999 9)、Y2=1.791 5×105X2-5.836 2×103(r2=0.999 9),线性范围分别为:进样量0.318 4 ~10.190 0 μg、0.142 2 ~4.550 0 μg。 表明淫羊藿苷、川续断皂苷Ⅵ进样量与峰面积之间均具有良好的线性关系。

图4 淫羊藿苷、川续断皂苷Ⅵ的HPLC 色谱图Figure 4 HPLC chromatograms of icariin and asperosaponin Ⅵ
2.2.6 精密度试验取批号为201801005 的补肝益肾颗粒,按“2.2.2”项下方法制备供试品溶液,按“2.2.4”项下方法测定,连续进样测定6 次,计算淫羊藿苷和川续断皂苷Ⅵ峰面积RSD 值分别为0.38%和0.46%,表明仪器精密度良好。
2.2.7 稳定性试验取批号为201801005 的补肝益肾颗粒,按“2.2.2”项下方法制备供试品溶液,按“2.2.4”项下方法,分别在0、2、4、8、12、24 h 进样测定。 计算得淫羊藿苷和川续断皂苷Ⅵ峰面积RSD值分别为0.88%和0.97%,表明供试品溶液在24 h内稳定性良好。
2.2.8 重复性试验取批号为201801005 的补肝益肾颗粒,按“2.2.2”项下方法制备供试品溶液,按“2.2.4”项下方法测定,平行操作6 份。 计算得淫羊藿苷和川续断皂苷Ⅵ的平均质量分数分别为0.148 8%、0.193 6%,RSD 值分别为1.60%和1.72%,表明本方法重复性良好。
2.2.9 加样回收率试验取已知淫羊藿苷、川续断皂苷Ⅵ质量分数分别为0.149 4%、0.193 0%的补肝益肾颗粒(批号:201801005)9 份,每份0.5 g,精密称定。 分别加入为样品中待测成分约0.5、1.0、1.5倍量的淫羊藿苷和川续断皂苷Ⅵ,按“2.2.2”项下方法制备供试品溶液,再按“2.2.4”项下色谱条件分别进样测定,计算回收率。 结果淫羊藿苷和川续断皂苷Ⅵ平均回收率分别为99.82%、100.70%,表明回收率良好。 见表1。
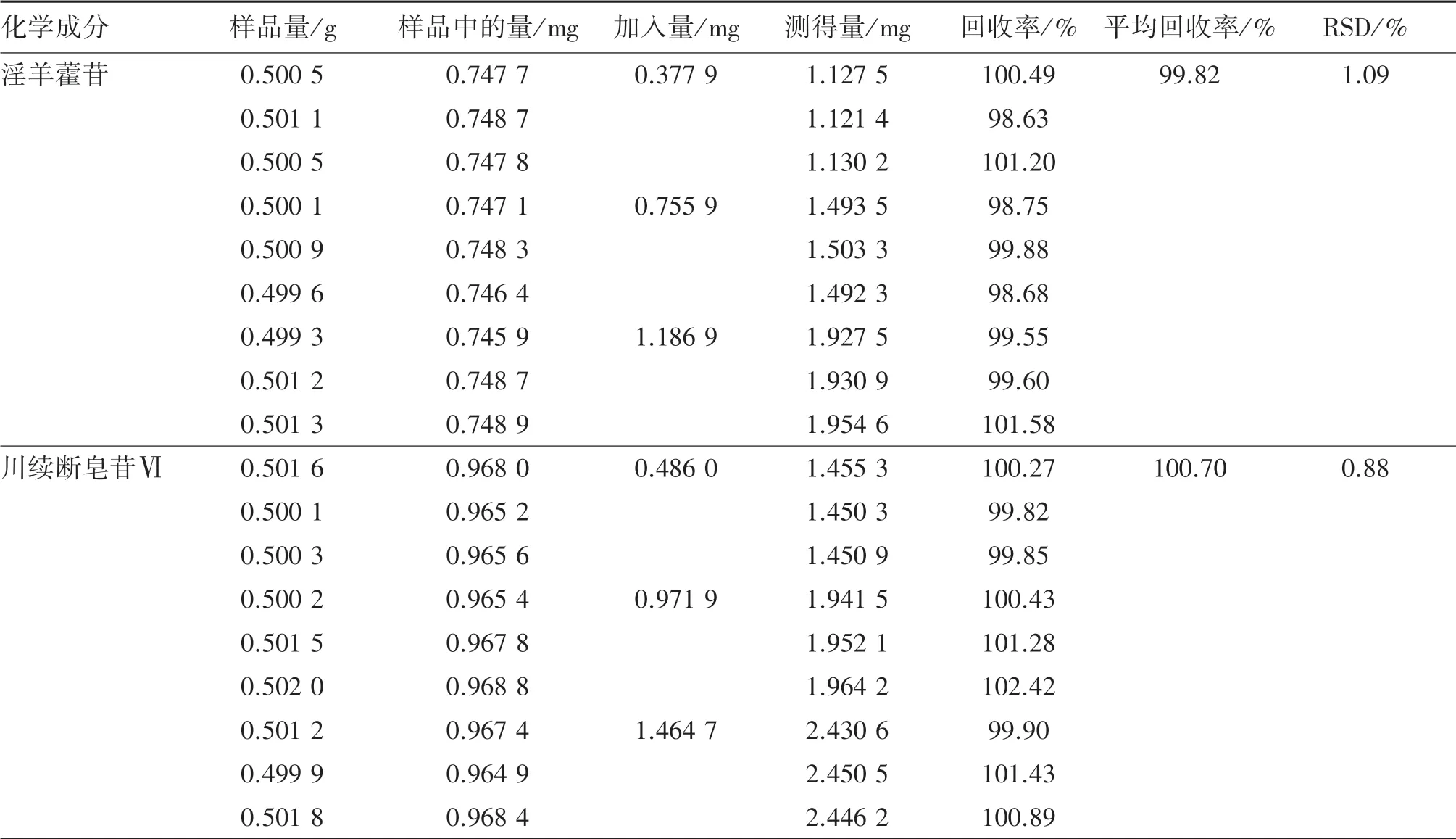
表1 补肝益肾颗粒中淫羊藿苷、川续断皂苷Ⅵ加样回收率试验结果Table 1 Recovery results of icariin and asperosaponin Ⅵin Bugan Yishen granules (n=9)
2.2.10 样品质量分数测定分别取批号20181004、20181005、20181006 的补肝益肾颗粒各2 份,按照“2.2.2”项下方法制备供试品溶液,再按“2.2.4”项下色谱条件进样测定,计算淫羊藿苷和川续断皂苷Ⅵ的质量分数,结果见表2。
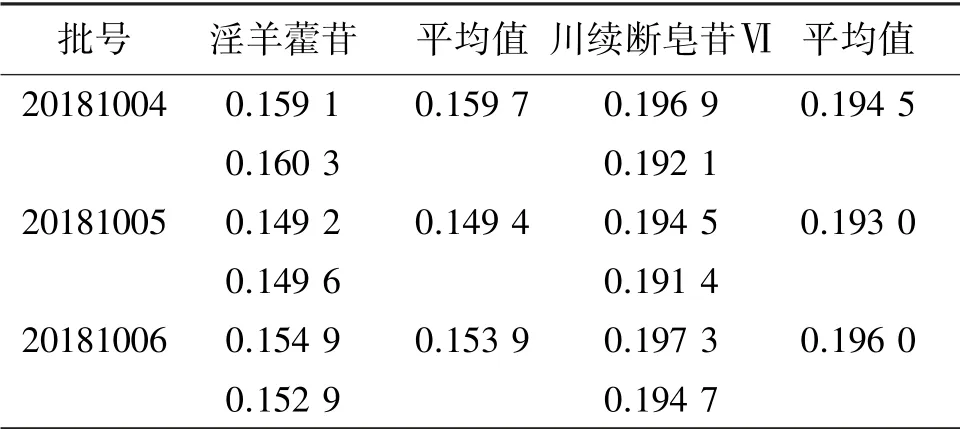
表2 补肝益肾颗粒中淫羊藿苷、川续断皂苷Ⅵ质量分数测定结果Table 2 The contents of icariin and asperosaponin Ⅵin Bugan Yishen granules (n=2) w/%
结果显示,3 批补肝益肾颗粒中淫羊藿苷、川续断皂苷Ⅵ的平均质量分数分别为0.154 3%和0.194 5%,以平均质量分数的80%设限,规定本品淫羊藿苷质量分数不得低于0.124%,川续断皂苷Ⅵ质量分数不得低于0.156%。
3 讨论
补肝益肾颗粒为水提制剂,因此进行质量分数测定样品前处理时考察了甲醇-水和乙醇-水溶媒体系,分别在甲醇、50%(体积分数,下同)甲醇、乙醇和50%乙醇4 种溶媒条件下测定淫羊藿苷和川续断皂苷Ⅵ的质量分数,结果表明甲醇提取效果最佳。淫羊藿苷最大吸收波长为270 nm,而川续断皂苷Ⅵ为212 nm,经试验发现,在212 nm 下测定淫羊藿苷质量分数与270 nm 下检测时相比,结果区别不大,因此选取212 nm 作为测定波长同时测定2 种化学成分的质量分数。
本试验建立的质量标准可快速、准确反映该制剂质量,为其临床疗效及质量控制提供了理论依据,对补肝益肾颗粒的进一步开发具有一定的指导意义。
