全程式中药安全性评价和监管
2020-03-16孙祖越王海学宋海波
韩 玲,孙祖越,杨 威,王海学,宋海波
〔1.国家药品监督管理局药品审评中心,北京 100038;2.上海市计划生育科学研究所,中国生育调节药物毒理检测中心,上海 200032;3.广东省药物非临床评价研究重点实验室(广东莱恩医药研究院有限公司),广东 广州 510990;4.国家药品监督管理局药品评价中心,北京 100038;5.香港科技大学生命科学部,香港 999077〕
中药的研发路径与化学药不完全相同,前者多数来源于临床实践,后者则常始于靶点和作用机制的发现。中药特别是中药复方或单味药的安全性大多源于临床发现,承于非临床,最后又回到临床,故称“全程式中药安全性评价”,即中药的安全性评价贯穿于从药物发现或临床人用经验到上市应用的全过程。
中药的监管也是如此,新版《药品管理法》[1]中有关主体责任和监管责任、全生命周期管理和监管有如此的表述。第一章《总则》中明确:“国家对药品管理实行药品上市许可持有人制度。上市许可持有人依法对药品研制、生产、经营、使用全过程中的安全性、有效性和质量可控性负责。”第二章《药品研制和注册》中明确:“国家建立药物警戒制度,对药品不良反应及其其他与用药有关有害反应进行监测、识别、评估和控制”,并明确了临床试验期间,发现存在安全性问题或者其他风险的,申办者和监管部门应负有的责任,如调整临床试验方案、暂停或终止临床试验等。第三章《药品上市许可持有人》中明确,上市许可持有人应对药品非临床研究、临床试验、生产经营、上市后研究、不良反应监测及报告与处理等承担责任。
这种“临床-非临床-临床-上市后研究”的循环,就是全生命周期或全程式的中药安全性评价和监管”的写真(图1)。
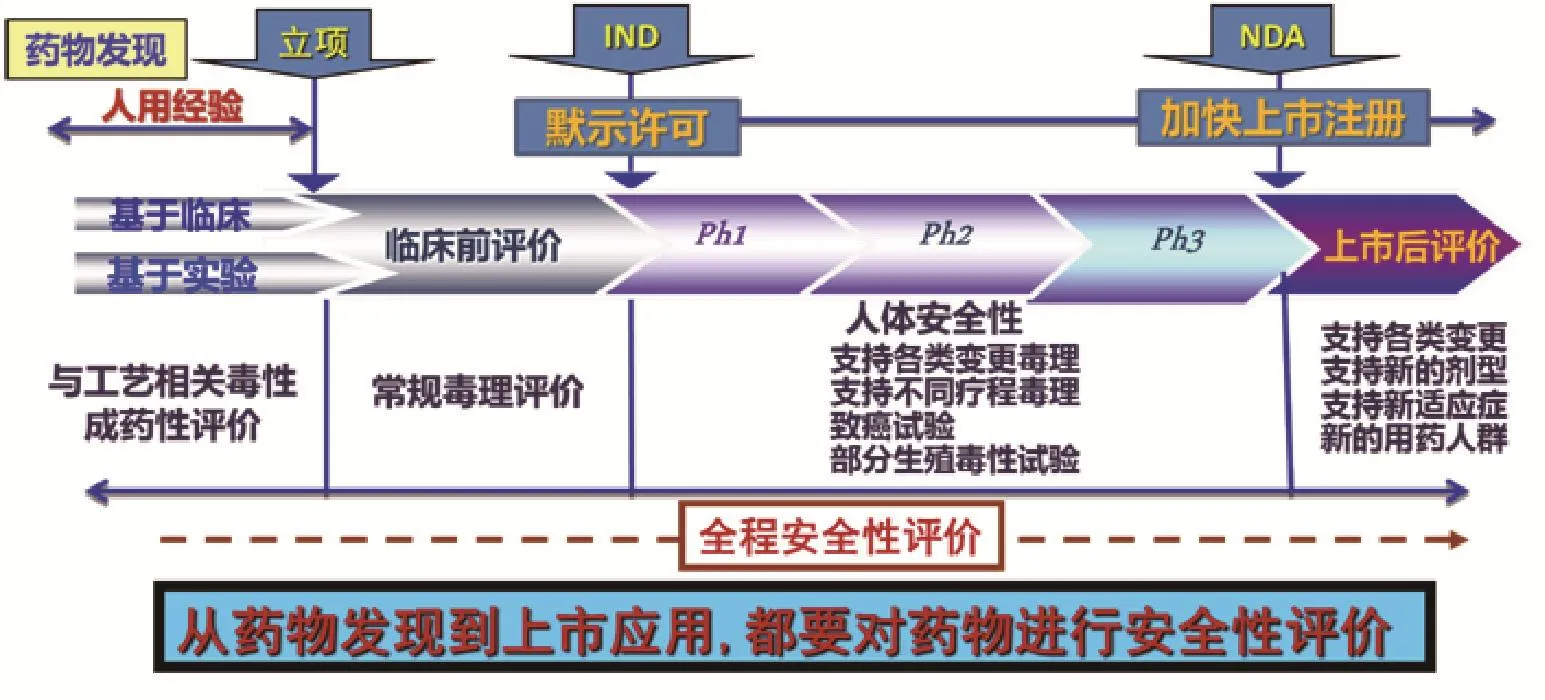
图1 安全性评价贯穿中药研发的全过程.IND:新药研究申请;NDA:新药申请;Ph1:Ⅰ期临床;Ph2:Ⅱ期临床;Ph3:Ⅲ期临床.
1 对中药毒性的认知源于临床经验
中药安全性评价来源于临床实践,来源于人们长期以来对中药毒性的认知,这是中药研发中的重要特点。
1.1 神农尝百草的传说由来已久
神农尝百草在发现中药功效的同时也开启了中药毒理学的研究,开创了药物毒性发现的先河[2]。东汉时期的著作《神农本草经》中记载了药物的“君臣佐使”及在方剂配伍中的地位和作用,药物的阴阳配合、七情和合、四气五味、有毒无毒,药物的采造,药物的煎煮方法,以及药物与病症的关系等临床用药法则,并沿用至今,指导着海内外炎黄子孙应用药物治疗疾病,保健强身。
1.2 中医文献古籍记载五彩斑斓
古代文献对中药毒性的记载,主要源于临床人用经验。例如,在《吴普本草》《名医别录》《新修本草》《重修政和经史证类备急本草》《本草纲目》和《中华本草》等[3]历代本草书籍中,均发现有毒中药的相关记载(表1)。
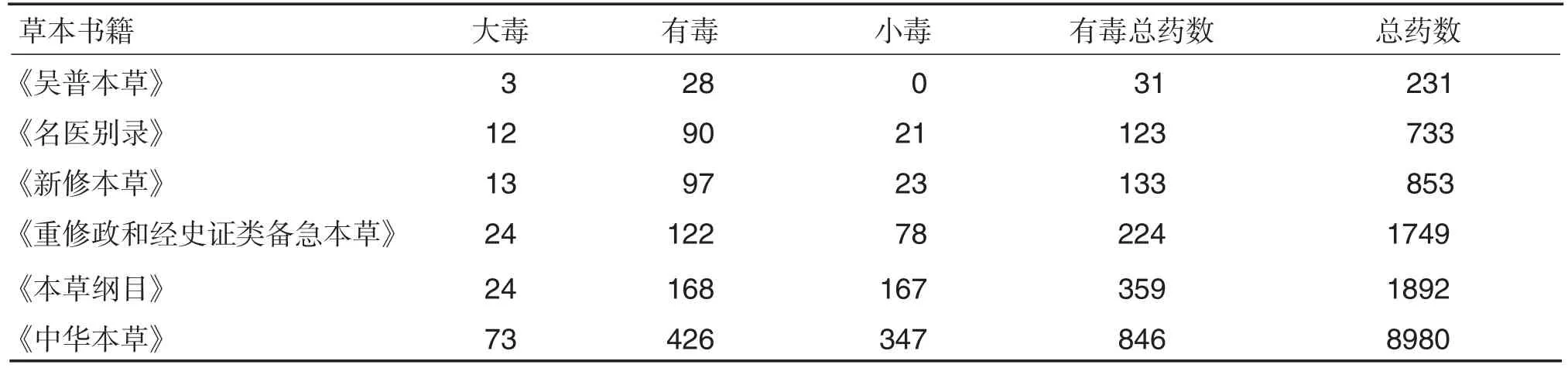
表1 中医文献古籍中有关中药安全性的记载
1.3 我国药典收录毒性药材种类繁多
《中华人民共和国药典》(2015年版)收录了83种毒性中药材及其饮片的毒性内容和临床使用中的注意事项,其中10种标注为“大毒”,42种“有毒”,31种“小毒”;还收录了孕妇禁用和慎用的药材与饮片总计99种,其中禁用39种,慎用60种(表2)[3-5]。

表2 《中华人民共和国药典》(2015年版)中收录的具有毒性的中药材及其饮片
由此可见,人们对中药毒性的认识先源于临床经验,来自广大的民间。
2 中药毒性评价循证于临床-非临床-临床-上市后再评价
一般而言,单味中药或复方应用的毒性报道首先来自于临床不良反应的观察,继而回到实验室开展深入系统的非临床毒性研究,获准后进入临床试验阶段,即使上市后还要深入开展上市后安全性再评价,这反映出中药毒性评价的普遍规律。
2.1 中药毒性在临床应用中时有报道
随着在临床上的广泛应用,中药临床不良反应的报道屡见不鲜,这些不良反应特别是严重的不良反应,会引发人们对中药安全性的关注和担忧。中药临床不良反应涉及全身各组织器官,严重不良反应甚至可能危及生命。例如,以双酯型生物碱为主要成分的乌头生物碱类中药,可引起心律失常、心悸、胸闷等心血管系统不良反应[6]。含砷中药雄黄可引起中枢神经系统细胞缺氧及功能紊乱,出现头晕、头痛、乏力等神经系统症状,严重时可出现抽搐、昏迷,甚至死亡[7]。蒽醌类成分如大黄素、大黄酸可引发肝损伤或肾损伤[8-9]。长期使用含马兜铃科酸的药材,可能引发急性或亚急性进行性、不可逆性肾损伤[10]。国家药品不良反应监测中心发布的药品不良反应通报中,涉及大量中药不良反应的内容。例如,第4期中发布了警惕鱼腥草注射液引起的不良反应,即鱼腥草注射液临床上用于清热、解毒、利湿,可引发过敏性休克、呼吸困难等严重的不良反应[11]。第22期中发布了警惕双黄连注射剂的严重不良反应,即具有清热解毒、疏风解表功效的双黄连注射剂是由金银花、黄芩、连翘提取物制备的中药制剂,可引发过敏性休克、过敏样反应、高热、寒战等全身性损害,呼吸困难、呼吸急促、喉水肿、支气管痉挛等呼吸系统损害,发疹型药疹、血管神经性水肿、剥脱性皮炎、重症多形性红斑等皮肤及其附件损害以及肝肾损伤等不良反应[12]。第61期中发布了关注口服何首乌及其成方制剂引起的肝损伤风险,口服何首乌及其成方制剂可能有引起肝损伤的风险,不良反应和(或)事件临床表现主要为全身乏力、食欲不振、厌油等消化道症状,尿黄、目黄、皮肤黄染等黄疸表现以及胆红素及转氨酶升高等实验室检查异常[13],等等。
2.2 动物实验发现中药潜在毒性
随着毒理学研究的不断发展,中药获批上临床试验之前,要求开展一系列的非临床安全性评价工作[14]。中药新药常规评价阶段毒性发现,主要包括含毒性药材复方的预期毒性发现、无毒性药材复方的非预期毒性发现、重复给药毒性试验发现的可疑致癌性或生殖毒性几类。其中含毒性药材复方的预期毒性发现,包括含大毒药材复方的毒性、含多味小毒药材复方的毒性、含重金属中药复方的毒性等几种情况。洋地黄、万年青、八角枫、蟾酥和夹竹桃等含强心苷类的药材能够增强心肌收缩,减慢心率,具有潜在的心脏毒性;木通、黄药子和商陆等含皂苷的中药材可能抑制呼吸,导致心、肾损害。某些中药复方制剂,尽管组方的药味均无毒性的记载或报道,但非临床评价中却出现明显的毒性,审评中发现某些常用中药组方制剂出现生殖毒性,已上市品种改变工艺后的复方出现原工艺没有的肝毒性,等等[15]。即在非临床研究中,出现预期毒性药材并未出现预期的较强毒性,而无毒性药材组方长期使用后却存在较大安全风险。例如,川乌水提物对痹证SD大鼠模型的生长发育和造血系统无明显毒性作用,在高剂量下表现出肝、肾和心脏毒性[16]。长期、大剂量使用被普遍认为无毒的中药如大黄和益母草等也有可能导致肾毒性发生。含有干扰核酸代谢、细胞分裂和细胞周期等成分的抗肿瘤中药,可能损伤正常细胞DNA引发细胞毒性。含马兜铃酸的复方制剂可能通过马兜铃酸A引发DNA加合物的形成,从而造成肝、肾细胞损伤,引起器官组织纤维化甚至肿瘤发生等[13]。
现代工艺与中药安全性也密切关联,选择工艺时需了解与临床实际用受试物工艺的一致性,并关注参考文献的提示作用或价值。采用现代工艺加工方式,特别是一些特殊工艺,如大孔吸附树脂、超临界CO2萃取和提取挥发油等,有时可能是产生中药毒性的原因。如含挥发油类的新药易出现溶血相关的毒性表现。以某中药为例,所含药味均为常用药材或饮片,但采用了非水煎工艺,动物的安全性研究出现明显的毒性(表3)[13]。
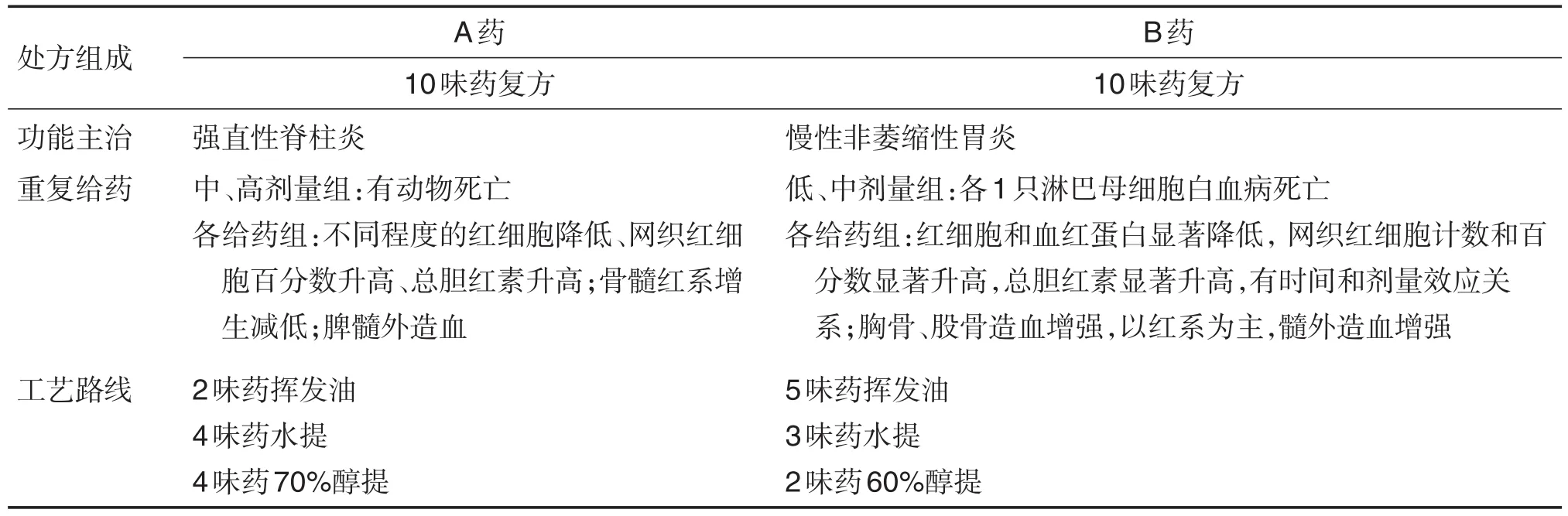
表3 工艺改变对动物血液系统影响的案例
2.3 临床期间继续进行的毒理学研究
临床期间继续进行的毒理学研究主要指针对临床批件内容支持后续临床分期的重复给药毒性试验,以及临床试验期间需继续完成的特殊毒性试验,如生殖毒性、遗传毒性和致癌试验等。临床期间若发现非预期的不良反应,需进行毒理学研究;临床期间发生的变更,如生产工艺的重大变更,需重新进行非临床安全性研究,并按照改良型新药进行申报;若临床试验方案变更,需增加用量或延长疗程的,应结合原非临床安全性试验结果,判断是否需提高剂量或延长给药周期重新进行毒理学试验;若临床期间拟新增适应证,应按照改良型新药重新申请IND;若拟增加用药人群,如增加儿童用药或降低儿童适用人群的年龄,也需要有支持其安全性评价的毒理学资料[17],按照补充申请申报等。
2.4 中药上市后再评价发现毒性
由于实验动物和人存在着种属差异,非临床毒理学研究时常无法全面评估出中药的不良反应,从而导致药品在临床试验阶段以及上市以后出现一些始料不及的毒性事件,或临床试验样本量较少不足以发现发生率较低的严重不良反应或扩大人群及范围后出现的非预期的不良反应等。因此,中药新药获批上市,并不代表就没有安全性问题了,还需要开展更广泛用药人群的上市后安全性再评价及毒性机制研究。如上市后再评价可结合新药出现的毒性进行毒性药材、毒性成分或复方制剂的毒理学基础研究,还有中药生殖毒性、肝毒性和肾毒性等研究。基于不良反应的毒理再研究或(和)再评价,发现一些中药毒性远超过非临床研究阶段的发现,则往往需要开展实验室的再评价,如马兜铃酸的肾毒性和致癌性研究、大黄蒽醌类致癌性研究、小檗碱心脏毒性的研究等均是药物上市后暴露出毒性进行再评价的结果[13]。《中药大辞典》收录的含马兜铃属药材有马兜铃、关木通、青木香、广防己、天仙藤、寻骨风、青香藤、南木香、通城虎、假大薯、淮通、管南香、鼻血雷、细辛、淫羊藿、何首乌和雷公藤等。文献报道的部分毒性药材及毒性成分毒理再研究和(或)再评价案例见表4。
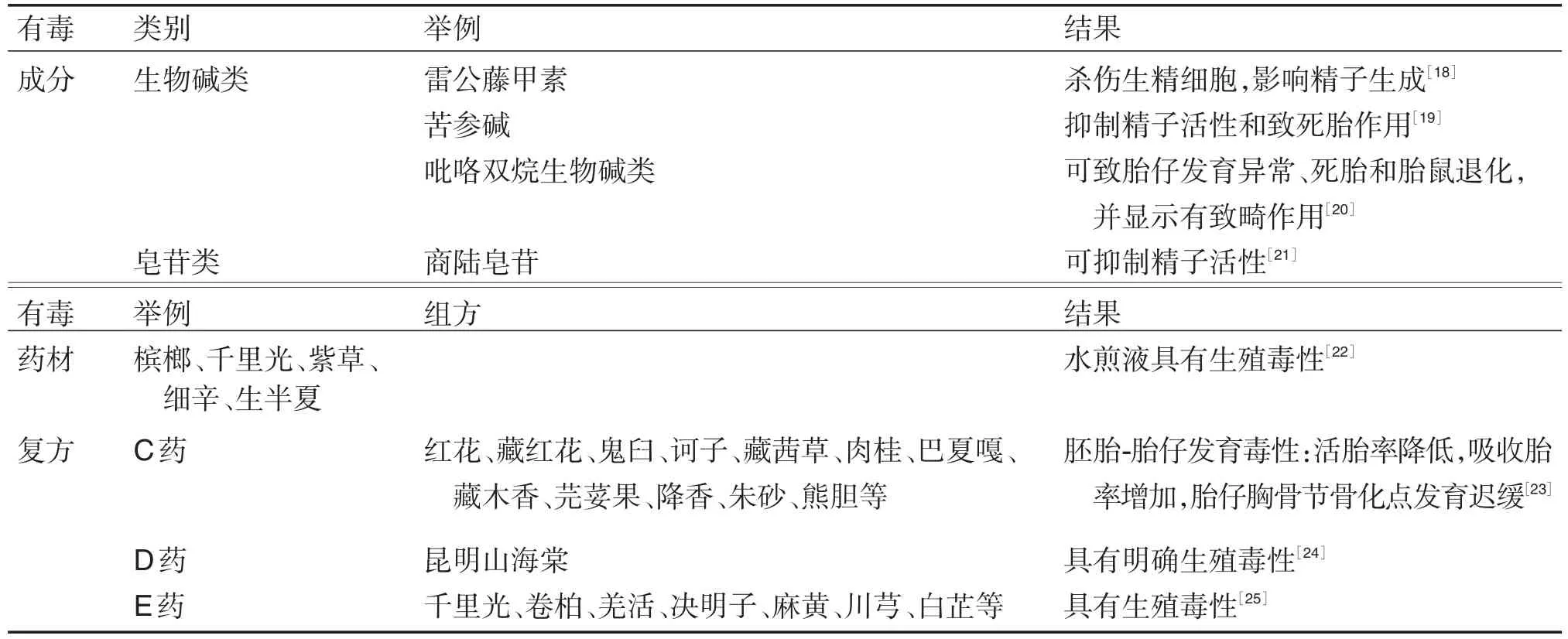
表4 毒性药材及毒性成分毒理再研究和(或)再评价举例
毒性基础研究对保证中药质量控制具有重要作用。周跃华等[18]针对大毒、剧毒、毒性较大(51种大毒药材)中药材国家标准中大毒药材的基础研究进行了总结。其中,只有16种(31%)已建立了毒性成分的含测项,3种(6%)已建立了指标成分的含测项,32种(63%)尚未建立任何成分的含测项。《中华人民共和国药典》(2015年版)收载的大毒药材标准的可控性相对较强,已针对其中22种中的14种建立了含量测定方法。但在地方药材标准中,大多毒性药材尚未建立与毒性相关的检测方法,标准的可控性较低,如在25种大毒药材中,仅雷公藤和雪上一枝蒿建立了毒性相关的检测方法[19]。
《医疗用毒性药品管理办法》(中华人民共和国国务院令第23号)[20]中将生马钱子、生川乌、生草乌、生巴豆、斑蝥、闹洋花、红粉、天仙子、生白附子、生附子、生半夏、生南星、生甘遂、生狼毒、生千金子、蟾酥、洋金花、雄黄、轻粉、砒石、砒霜、水银、青娘虫、红娘虫、生藤黄、生雪上一枝蒿、红生丹和白降丹列入毒剧药管理的28种中药材(大毒药材)。汇总见表5。
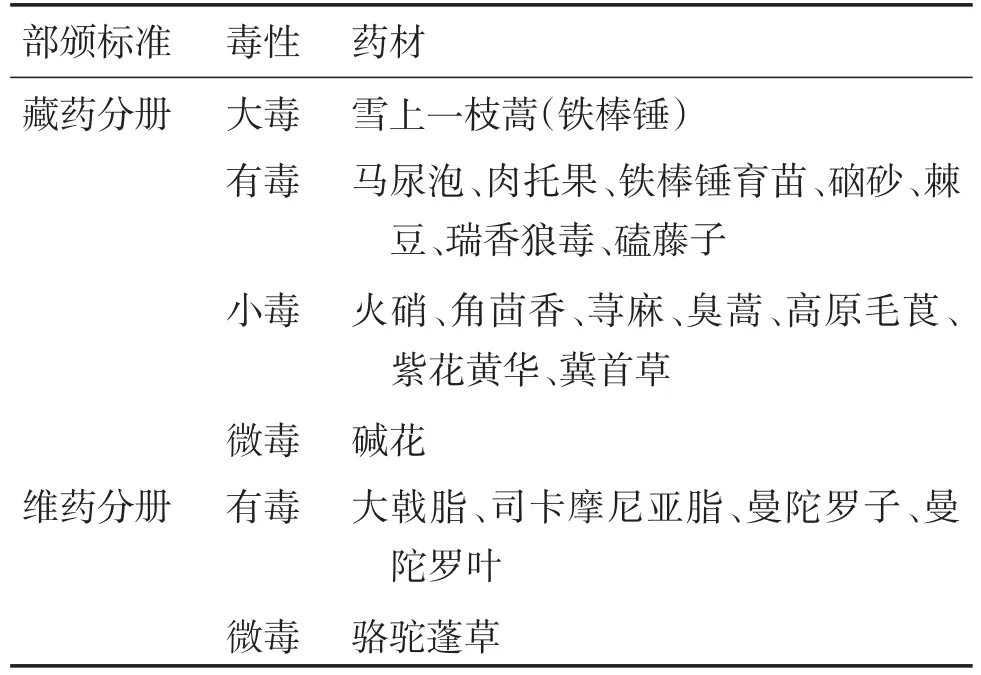
表5 部颁标准中有关中药毒性药材
2.5 中药不良反应监测堪称重中之重
中药临床不良反应是中药安全性评价全过程的重要组成部分和主要评价依据。根据国家药品不良反应监测中心发布的《国家药品不良反应监测年度报告(2019)》[21],2019年累计收到《药品不良反应/事件报告表》151.4万份,涉及怀疑药品163.5万例次,其中中药占12.7%。涉及《国家基本药物目录》(2018版)中成药共268个品种,不良反应/事件报告8.9万例次,其中严重报告6 692例次(7.6%)。2019年国家基本药物中成药部分7大类中,药品不良反应/事件报告总数由多到少依次为内科用药、骨伤科用药、妇科用药、外科用药、耳鼻喉科用药、儿科用药、眼科用药。涉及怀疑药品19.9万例次,其中中药占7.1%。按照剂型分类,整体报告中注射剂占63.3%,严重报告中注射剂占74.3%。所有注射剂报告中,中药注射剂占9.1%。中药不良反应发生排名前5位的类别分别是理血剂中活血化瘀药(28.4%),清热剂中清热解毒药(11.4%),补益剂中益气养阴药(6.8%),开窍剂中凉开药(6.1%),祛湿剂中清热除湿药(5.7%)。严重不良反应/事件报告的例次数排名前5位的类别分别是理血剂中活血化瘀药(39.8%),补益剂中益气养阴药(13.0%),开窍剂中凉开药(10.5%),清热剂中清热解毒药(8.6%),解表剂中辛凉解表药(3.8%)。按照给药途径分布,注射给药占45.5%,口服给药占46.4%,其他给药途径占8.1%。注射给药中,静脉注射给药占98.5%,其他注射给药占1.5%。中药不良反应的常见因素主要包括“与功效相关的不良反应”、“与指标成分相关的不良反应”、“与辅料相关的不良反应”、以及“与溶媒相关的不良反应”几类。重视毒性中药的不良反应监测管理,依托现有规范化的药品不良反应监测与报告体系,正确认识并对待毒性中药不良反应,为毒性中药科学合理应用提供依据和对策,有助于中药现代化以及全球化。
总体而言,中药获批上市并不是安全性评价的终点,而是安全性评价的重新开始,特别是研究薄弱的特殊毒性更是要开展上市后的安全性再评价,如生殖毒性、遗传毒性和致癌试验等。还有特殊用药人群更不可忽视,如儿童和老年人用药等。
3 药物警戒应贯穿于药品全生命周期
3.1 药物警戒概念的变化
1974年药物警戒(pharmacovigilance,PV)概念在法国问世,着重强调药物不良反应(adverse drug reaction,ADR)的监测。2002年,WHO重新对PV进行了定义[22],指出PV不仅涉及药物ADR,还涵盖其他所有与药物相关的科学研究与活动,即为一系列发现、评价、认识和预防药物不良作用或其他任何与药物相关问题的科学和活动。PV的涵盖范围从原来的上市后安全信号的收集、识别、分析、评估和风险管理延展到药物的研发阶段。国际医学组织理事会(Council for International Organi⁃zations of Medical Sciences,CIOMS)在2005年制定并发布的CIOMSⅥ《临床试验安全信息管理》[23]中明确,PV也包括上市前(临床试验期间)安全信息的收集、监测和评估;风险识别、风险评估和风险最小化概念也适应于上市前药物。
3.2 药物警戒执行国际通行标准
我国临床试验期间PV制度与技术体系建设起步较晚。2017年6月1日,中国正式加入国际人用药品注册技术协调委员会(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)成员国,加速了药品监管与国际全面接轨的进程。
2018年1月25日,原国家食品药品监督管理总局发布了《关于适用国际人用药品注册技术协调会二级指导原则的公告》[24](2018年第10号),要求“自2018年5月1日起,药物临床研究期间报告严重且非预期的药品不良反应适用《E2A:临床安全数据的管理:快速报告的定义和标准》《M1:监管活动医学词典(Med DRA)》和《E2B(R3):临床安全数据的管理:个例安全报告传输的数据元素》”。
2018年7月在原国家食品药品监督管理总局发布《关于调整药物临床试验审评审批程序的公告》[25](2018年第50号)中明确要求:① 对于药物临床试验期间出现的可疑且非预期严重不良反应和毒理研究提示重大安全性风险信号,申请人应按照《药物临床试验期间安全性数据快速报告标准和程序》中相关要求向药品审评中心(Center for Drug Evaluation,CDE)递交(个例)安全性报告。药审中心可根据审查需要,要求申请人修改临床试验方案,必要时暂停临床试验。②申请人在获得首次临床试验许可后,应定期向CDE提供药物研发期间安全性更新报告。CDE可根据审查情况,要求申请人调整报告周期。逾期未提交的,申请人应暂停药物临床试验[26]。
在2020年新修订的《药品管理法》[1]中明确国家建立PV制度,以及申办者及药品监督管理部门应当承担的责任和义务。
在2020年7月1日起实施的新版《药品注册管理办法》[27]第二十八条中也提出申办者应当定期在CDE网站提交研发期间安全性更新报告;对于药物临床试验期间出现的可疑且非预期严重不良反应和其他潜在的严重安全性风险信息,应当及时报告等内容。根据安全性风险严重程度,申办者应采取药监部门依职责可以责令调整临床试验方案、暂停或者终止药物临床试验。
为落实上述法律法规及公告要求,自2018年初,CDE开展了PV体系建设和标准的研究制定工作,发布了系列相关的技术文件和规范。2018年4月27日发布的《药物临床试验期间安全性数据快速报告标准和程序》[28]中明确了我国药物临床试验期间可疑且非预期严重不良反应(Suspected Unex⁃pected Serious Adverse Reaction,SUSAR)快速报告的目的、范围、内容、时间与时限、报告途径与方法、报告责任主体和监管主体等相关要求。2018年7月30日发布的《E2B(R2)安全性消息处理和个例安全性报告技术规范》[29]中对个例安全性报告数据的报告明确了细则要求。2020年7月1日发布的《药物临床试验期间安全信息评估与管理规范(试行)》[30]和《研发期间安全性更新报告管理规范(试行)》[31],明确了药物临床试验期间安全风险管理中申请人的主体责任、监管机构的监督责任以及相关工作程序;明确了研发期间安全性更新报告(Devel⁃opment Safety Update Report,DSUR)内容、格式、报告途径和相关程序等方面的要求。
3.3 临床试验期间的风险控制
申请人(申办者)是药物临床试验安全风险管理的主体责任人。申请人(申办者)在临床试验过程中应严格遵守相关法律法规,确实承担起主体责任。
中药新药临床试验过程也有出现严重安全性风险的案例。如2008年5月原国家食品药品监督管理总局收到正在进行的6类复方制剂“仙牛健骨颗粒”Ⅲ期临床试验期间发生的严重不良事件“急性肝损伤”[32]的报告。原国家食品药品监督管理总局立即启动了全面调查。4批临床用样品急性毒性试验结果表明,动物死亡率分别为60%(1次·d-1),95%,80%和75%(2次·d-1),但该药在申请临床时的研究中并未见任何急性毒性数据支持。但仅根据急性毒性试验结果尚无法判断该药是否具有肝毒性,尚需进行重复给药毒性试验。临床试验检查发现主要问题如下:①临床试验方案在安全性方面存在严重缺陷,6个月的疗程仅设计了用药前后各1次的安全性检查,且肝功能指标不全面,如缺胆红素指标;在Ⅱ期临床试验已有1例发生严重肝损伤的情况下,Ⅲ期方案在安全性方面并未作任何修改。②Ⅲ期临床试验的知情同意书内容过于简单,未告知受试者任何可能的不良反应,未描述Ⅱ期试验中出现肝损伤的情况。③伦理委员会也未要求申办者和研究者修改Ⅲ期方案,增加反映肝功能损害的相应指标。且在Ⅲ期试验发生多起严重肝损伤的严重不良事件后,也未采取有力的干预措施,如召回受试者进行安全性检查和及时暂停临床试验等。原国家食品药品监督管理总局根据检查结果综合考虑,认为发生的严重不良事件与药物自身毒性有明确相关性,最终责令终止了该临床试验。
虽然此次查明该严重不良事件系药物所致,该药处方由淫羊藿、补骨脂和杜仲等7味药组成,尚无更多数据支持可找到与肝毒性直接相关的原因,需要深入研究。此外,在研究中发现对照药也出现了明显的不良反应,因此建议应对已上市对照药物进行再评价。
通过该案例,说明中药临床试验期间的安全性不容忽视,同时也引发了我们对中药临床研究及监管工作中诸多问题的思考。
3.4 临床期间变更的风险控制
依据《药品注册管理办法》[31],在药物临床试验期间,发生药物临床试验方案变更、非临床或者药学的变化或者新发现,申请人应当充分评估对受试者安全的影响。申请人评估认为不影响受试者安全的,可直接实施并在药物DSUR中报告。申请人评估认为可能增加受试者安全性风险的,应当向CDE提出补充申请。
建议关注《药品注册管理办法》相关配套文件《药物临床试验登记与信息公示管理制度》《药物临床试验期间药物研发期间安全性更新报告要求及管理规定》《药物临床试验过程中一般风险管控及责令暂停、终止工作程序》等。
3.5 上市后不断完善说明书安全性内容
《中医药发展战略规划纲要(2016-2030年)》[33]中指出,“开展中成药上市后再评价,加大中成药二次开发力度,开展大规模、规范化临床试验,培育一批具有国际竞争力的名方大药”。6万个已批准上市的中药制剂批准文号仅有15%~35%的批准文号有生产上市信息,约2000个中药制剂品种处于休眠状态(无厂家生产)。部分中成药功能主治描述宽泛且临床支持依据不足,迫切需要对此类中成药进行安全性和有效性的再评价,有依据的修订功能主治,完善精准的临床用药定位。由于病例入选数量及标准限制,Ⅰ~Ⅲ期临床研究有效性和安全性研究存在一定局限性,且无法提供更多实际临床用药中的药物反应,如患者年龄、机体状态、药物相互作用等因素。许多说明书中安全项内容至今仍为“尚不明确”。这些缺课内容需要及时补充完成,需要申请人及监管部门重视,并不断推进中药的再评价工作。
药品说明书在新药研究的整个生命周期中应处于动态更新的过程。应根据非临床安全性数据、临床试验的发现、以及上市后的基础研究和不良反应发现持续进行更新和完善。以某新药为例,自2010年11月FDA批准上市后,原版说明书中关于生殖毒性信息处于不断更新状态;2012年3月第二版以及2013年6月第三版说明书内容中陆续增补了胚胎-胎儿毒性相应内容;2014年2月国内新药研究申请时的申报材料中增加了关于“雄性、雌性食蟹猴的生育力和早期胚胎发育”的相应内容;2018年6月最新版本说明书中增补了临床诊断的妊娠中,重大出生缺陷和流产的背景风险的相关信息(表6)。

表6 药物警戒中的某新药说明书生殖毒性信息更新
3.6 关注上市后变更的安全性
《药品注册管理办法》中有关药品上市后的变更内容包括:上市许可持有人应当主动开展药品上市后研究,对药品的安全性、有效性和质量可控性进行进一步确证,加强对已上市药品的持续管理。《药品注册批件》及附件要求持有人在药品上市后开展相关研究工作的,如上市后完成大样本量的安全性评价、完成相应的生殖毒性研究,针对毒性及作用机制进行探索,完成附条件批准的相应要求等等,持有人应当按照批件要求在规定时间内完成并按要求申报补充。此外,应根据《已上市中药变更及申报资料》的相应情况,按照其对药品安全性、有效性和质量可控性的风险和产生影响的程度,分为审批类变更、备案类变更和报告类变更3类。分清生产过程中的重大、中等或微小变更,确定是属于中药改良型新药,还是按照补充申请申报。按照说明书中涉及有效性内容的变更或补充完善安全性内容的变更,均有相关要求。药品说明书是临床合理用药的主要参考依据,应结合上市后应用及不良反应等持续监测,不断完善说明书中不良反应、注意事项、禁忌、特殊用药人群、药物相互作用、用量用法以及用药周期等与临床用药安全性相关的内容。国家药品监督管理局也动态监测上市后的不良反应情况,如2020年以来陆续发布双黄连颗粒等口服制剂、川贝枇杷制剂、黄连上清制剂、复方三七补血胶囊、麝香保心丸、鼻炎康片、心脑康制剂等药品说明书修订的公告,增加或完善了说明书中的警示语、不良反应、注意事项和禁忌等安全性信息。
事实证明,加强药品全生命周期的安全性监管尤为重要,除了一些毒性或不良反应能够在新药研发的各个阶段中发现,但尚有较多的毒性或不良反应是在非临床研究、局限性的临床试验以及上市后应用过程中未能被发现或被忽略的。
4 结语
综上所述,中药源于临床实践,最后又回到临床,有基本的临床个体或群体用药经验及安全性信息,又经过系统规范的非临床安全性评价和临床试验,到最终作为中成药上市,期间安全性评价应始终贯穿于全过程。针对中药的特殊性,安全性风险管理应贯穿于中药材的种植和溯源、炮制和加工、生产工艺和全过程质控,也要体现在系统规范及有针对性的毒理学研究、规范和风险可控的临床试验以及上市后的再评价的全过程之中。目前,针对源于中药临床实践的人用经验的总结、非临床安全性评价及临床试验期间和上市后的PV与风险监管制度及体系,均已建立了沟通交流制度和评价体系。这一过程应体现申请人或上市许可持有人的主体责任,不仅需要研发机构、安全性评价机构本着为用药人群安全负责的科学评价,提升安全管理意识和能力,同时也需要监管部门切实履行监管职责,切实保护好受试者和患者的安全。
