基于指纹图谱分析和多成分同时定量的玉屏风丸质量评价
2020-03-15王巧清
王巧清
(中国人民解放军联勤保障部队第900 医院,福建 福州 350000)
玉屏风丸由黄芪、炒白术及防风3 味中药材加工而成,具有益气、固表、止汗功效,主要用于表虚不固、自汗恶风,面色白,或体虚易感风邪者,现收载于《卫生部药品标准·中药成方制剂(第一册)》[1]。目前,相关研究多集中于玉屏风颗粒剂及散剂等的质量控制[2-3]。作为中医药经典剂型,丸剂具有代表性和传承性,玉屏风丸在胃肠道中溶散缓慢,药效持久,发挥药效“缓而不迟”,且方便服用、携带和贮存。复方药效物质基础及质量控制研究在于多味药材的多成分含量测定,而中药指纹图谱正是利用其“整体性”及“模糊性”的特点,全面反映中药化学的相关性,整体评价其质量。中药指纹图谱具有同时、高效、定性定量分析的特点,已广泛运用到中药复方多类多成分研究领域[4-6]。为了更全面、有效地评价玉屏风丸的质量,本研究中采用高效液相色谱(HPLC)法建立了玉屏风丸指纹图谱,并结合聚类分析,对各色谱峰所代表的化学成分进行了初步归属,建立同时测定其7 个主要成分指标(毛蕊异黄酮葡萄糖苷、芒柄花素、升麻素苷、升麻素、白术内酯Ⅲ、白术内酯Ⅱ、白术内酯Ⅰ)的定量检测方法,以期为系统和准确评价其质量提供技术参考。现报道如下。
1 仪器与材料
1.1 仪器
Waters 2695 型高效液相色谱仪,包括2695 型四元梯度泵、2695 型自动进样器、2489 型紫外可见光检测器、Empower3 色谱工作站,美国沃特世公司);AUW-220D 型电子天平(日本岛津公司,精度为0.01 mg);KQ-250DB 型数控超声波清洗器(昆山超声波仪器有限公司,功率为500W)。
1.2 试药
毛蕊异黄酮葡萄糖苷(calycosin7-O-β-D-glucopyranoside,批号为111920-201606,含量97.6%),芒炳 花 素(formononetin,批 号 为111703-201504,含 量100%),升麻素苷(prim-O-glucosylcimifugin,批号为111522-201712,含量96.2%),升麻素(cimifugin,批号为 111710-200602,含量 95.5% ),白术内酯Ⅲ(atractylenolide Ⅲ,批号为 111978-201501,含量99.9% ),白术内酯Ⅱ(atractylenolide Ⅱ,批号 为111976-201501,含量 99.9% )、白术内酯Ⅰ(atractylenolide Ⅰ,批 号 为 111975-201501,含 量99.9%),各对照品均购自中国食品药品检定研究院;乙腈、甲醇(美国Merck 公司)均为色谱纯;磷酸为分析纯;水为娃哈哈纯净水。玉屏风丸(市售,14 批,编号为S1 ~S14,S1 ~S10 为山西华康药业股份有限公司生产,规格为6 g×10 袋/盒,批号分别为20171212,20180105,20180207,20180313,20180408,20180518、20180626,20180912,20181019,20181115;S11 ~S14 为厦门福满药业有限公司生产,规格为6 g×18 袋/盒,批号分别为180906,181022,181108,181201)。
2 方法与结果
2.1 色谱条件[7 -9]
色谱柱:Shim-pack VP-ODS 柱(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-0.3%磷酸溶液(B),梯度洗脱(0 ~20 min 时12%A,20 ~40 min 时12%A→35%A,40 ~46 min 时35%A→68%A,46 ~50 min 时68%A→80%A,50 ~60 min 时80%A→12%A);波长:300 nm(0 ~2 min,升麻素苷),254 nm(20 ~40 min,毛蕊异黄酮葡萄糖苷、芒柄花素及升麻素),220 nm(40 ~46 min,白术内酯Ⅲ及白术内酯Ⅱ),275 nm(46 ~60 min,白术内酯Ⅰ);柱温:35 ℃;流速:0.85 mL/min;进样量:10 μL。
2.2 溶液制备
分别取毛蕊异黄酮葡萄糖苷、芒柄花素、升麻素苷、升麻素、白术内酯Ⅲ、白术内酯Ⅱ、白术内酯Ⅰ对照品各适量,精密称定,以85%甲醇溶解并定容,制成质量浓度分别为0.786 6,0.411 2,0.434 7,0.310 8,0.379 5,0.264 5,0.200 3 mg/mL 的混合对照品溶液。取装量差异项下的样品内容物,研细,取粉末约3.0 g,精密称定,置50 mL 棕色容量瓶中,加入85%甲醇溶液35 mL,室温超声处理(功率为450 W,频率为40 kHz)45 min 后取出,放冷,用85%甲醇溶液稀释至刻度,摇匀,滤过,即得供试品溶液。均室温避光保存。
2.3 指纹图谱的建立及分析
2.3.1 方法学考察
精密度试验:取同一批(批号为20180912)样品,按
2.2 项下方法制备供试品溶液,按拟订色谱条件连续进样6 次,记录色谱图。结果以毛蕊异黄酮葡萄糖苷为参比峰,计算得各相对峰面积的RSD均小于0.8%(n=6),共有峰相对保留时间的RSD均小于0.6% (n=6),表明仪器精密度良好。
重复性试验:取同一批(批号为20180912)样品,按2.2 项下方法平行制备供试品溶液6 份,按拟订色谱条件进样测定,记录色谱图。结果以毛蕊异黄酮葡萄糖苷为参比峰,计算得各相对峰面积的RSD均小于1.2%(n=6),共有峰相对保留时间的RSD均小于0.9%(n=6),表明方法重复性良好。
稳定性试验:取同一批(批号为20180912)样品,按2.2 项下方法制备供试品溶液,按拟订色谱条件分别于0,4,8,12,16,20,24 h 时进样,记录色谱图。结果以毛蕊异黄酮葡萄糖苷为参比峰,计算得各相对峰面积的RSD均 小 于1.0% (n=7),共 有 峰 相 对 保 留 时 间 的RSD均小于0.7% (n=7),表明玉屏风丸供试品溶液在24 h 内稳定。
2.3.2 指纹图谱的建立及相似度评价
按2.2 项下方法制备14 批供试品溶液,按拟订色谱条件进样测定,记录色谱图。结果见图1 至图3。将14份指纹图谱依次导入《中药色谱指纹图谱相似度评价系统》(2012A 版)软件,以S4 号玉屏风丸供试品溶液的指纹图谱作为参照图谱,设定时间窗宽度为0.3,采用多点校正全谱匹配,生成指纹图谱共有模式,并计算各图谱与共有模式的相似度。结果14 批样品的指纹图谱中,共得到18 个共有峰,14 批指纹图谱的相似度分别为0.994,0.995,0.999,0.998,0.997,0.996,0.991,0.993,0.992,0.995,0.989,0.979,0.988,0.990,均 大 于0.97。

图1 14 批样品高效液相色谱叠加指纹图谱

图2 玉屏风丸高效液相色谱对照品指纹图谱
2.3.3 部分共有峰指认及相对峰面积计算
将指纹图谱与2.3.2 项下混合对照品溶液色谱图进行比对,以保留时间为评价指标,指认出共有峰中7 种化合物,分别为4 号(升麻素苷)、9 号(毛蕊异黄酮葡萄糖苷)、10 号(升麻素)、11 号(芒柄花素)、16 号(白术内酯Ⅲ)、17 号(白术内酯Ⅱ)和18 号(白术内酯Ⅰ),其保留时间 平 均 值 分 别 为13.18,28.04,30.11,31.96,44.02,45.89,47.95 min。其中9 号峰信号最强,位置居中,选为参照峰,计算各共有峰的相对峰面积。结果见表1。
2.4 含量测定
2.4.1 方法学考察
线性关系考察及检测限和定量限测定:分别精密吸取2.2 项下混合对照品溶液1,2,5,10,15,20 μL,按拟订色谱条件进样测定,记录色谱图。以各组分对照品进样量(X,μg)为横坐标、峰面积(Y)为纵坐标进行线性回归,绘制标准曲线,计算各组分线性回归方程及相关系数。取2.2 项下混合对照品溶液,用85%甲醇逐步稀释,按各组分色谱峰信噪比(S/ N)=3 ∶1 及10 ∶1 分别测定其检测限(LOD)和定量限(LOQ)。结果见表2。
精密度试验:取混合对照品溶液,按拟订色谱条件连续进样6 次,记录峰面积。结果见表2,表明仪器精密度良好。
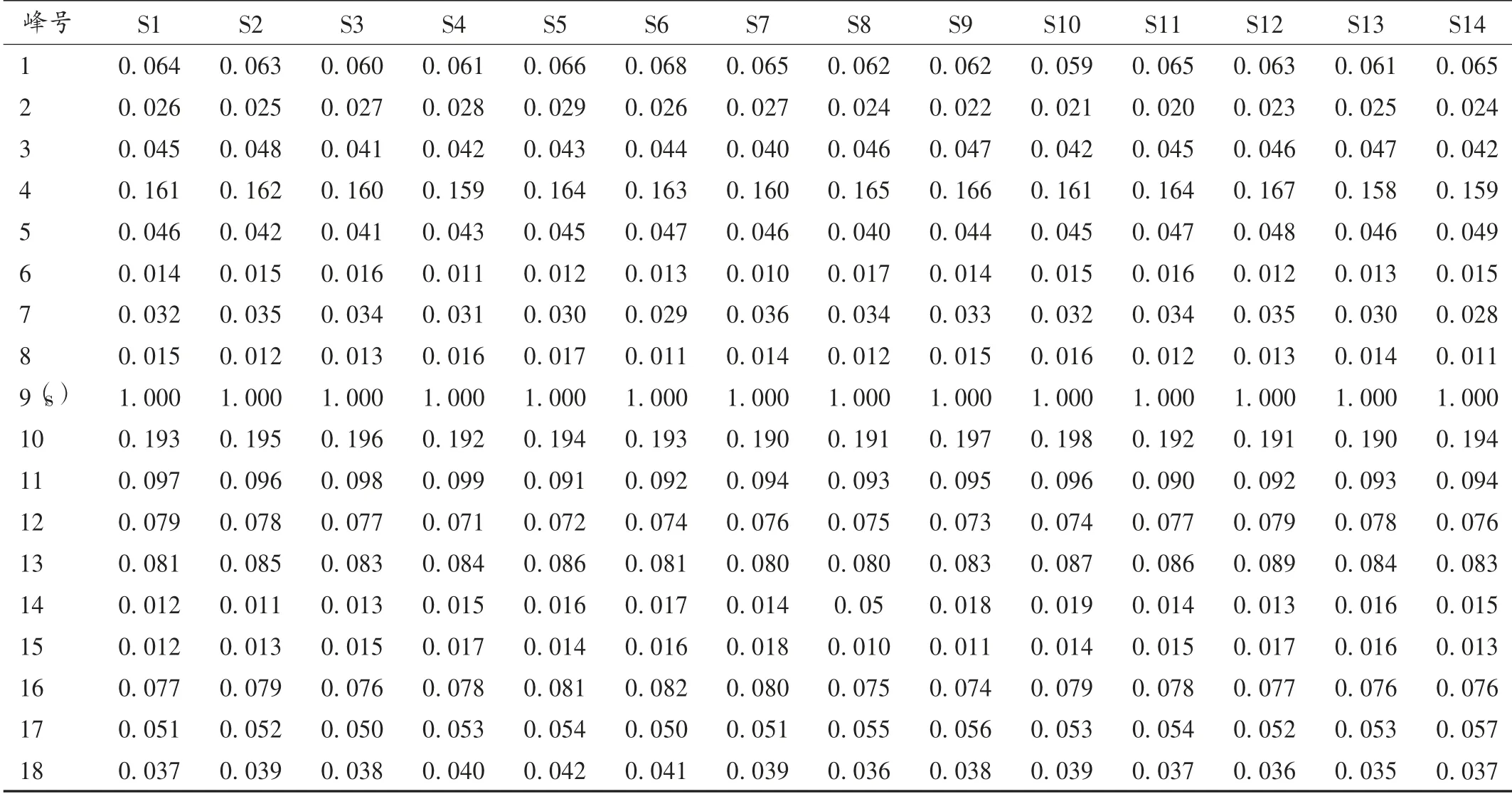
表1 14 批玉屏风丸(S1 ~S14)共有峰的相对峰面积
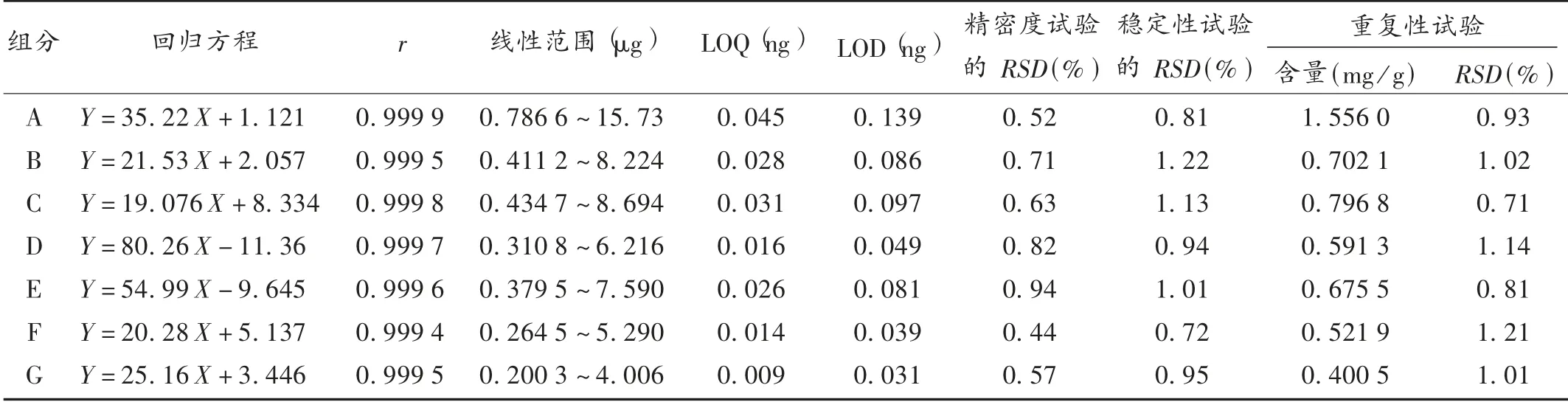
表2 玉屏风丸中7 个组分含量测定方法学考察结果
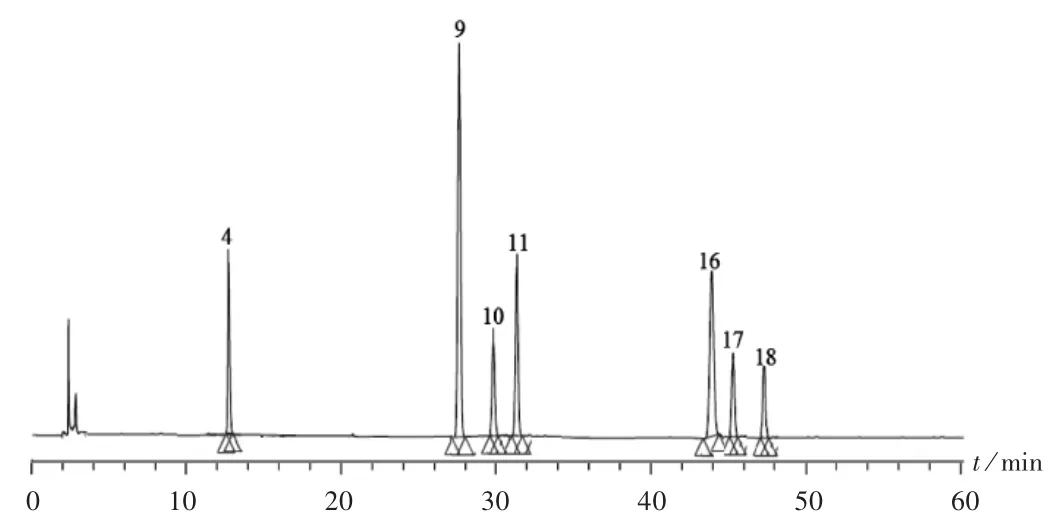
图3 混合对照品溶液高效液相色谱图
稳定性试验:取同一批(批号为20180912)样品,按2.2 项下方法制备供试品溶液,按拟订色谱条件分别于0,3,6,9,12,18,24 h 时进样,记录峰面积。结果见表2,表明供试品溶液在24 h 内稳定。
重复性试验:取同一批(批号为20180912)样品6 份,按2.2 项方法平行制备供试品溶液,按拟订色谱条件进样测定,记录峰面积。结果见表2,表明方法重复性良好。
加样回收试验:取同一批(批号为20180912)已知含量玉屏风丸粉末6 份,每份1.5 g,精密称定,置具塞棕色容量瓶中,精密加入含毛蕊异黄酮葡萄糖苷1.214 7 mg/mL、芒柄花素0.310 9 mg/mL、升麻素苷0.341 1 mg/mL、升麻素0.235 3 mg/mL、白术内酯Ⅲ0.290 6 mg/mL、白术内酯Ⅱ0.199 9 mg/mL 和白术内酯Ⅰ0.154 7 mg/mL的混合对照品溶液2.0 mL,按2.2 项下方法制备供试品溶液,摇匀,滤过,按拟订色谱条件进样测定,记录峰面积,计算各组分加样回收率。结果见表3。
2.4.2 样品含量测定
取S1 ~S14 号玉屏风丸,按2.2 项下方法制备供试品溶液,按拟订色谱条件进样测定,记录色谱图,按外标法分别计算各成分的含量。结果见表4。
2.5 系统聚类分析[10 -11]
系统聚类分析作为一种统计分析方法,是在相似性的基础上收集数据来分类。以14 批样品中18 个共有峰的相对峰面积为变量,采用SPSS 22.0 软件聚类法中Ward 方法对样品进行系统聚类分析,采用组间平均数联结法,以欧氏距离平方为测度。结果见图4。可见,当类间距为10 ~25 时,14 批样品可聚为两大类,第一类为S3,S9,S4,S5,S1,S6,S8,S2,S10,S7;第二类为S12,S14,S11,S13。第一类均为山西华康药业股份有限公司产品,第二类均为厦门福满药业有限公司产品,说明同一厂家产品的质量稳定性较高。
3 讨论
3.1 测定目标成分选择
黄芪作为玉屏风丸的君药,占总处方量的60%,具有利水消肿、行滞通痹、托毒排脓、敛疮生肌之功效,来源于豆科植物蒙古黄芪Astragalus membranaceus(Fisch.)Bge.var.mongholicus(Bge.)Hsiao 或膜荚黄芪Astragalus membranaceus(Fisch.)Bge. 的干燥根,主要活性成分为黄酮类(毛蕊异黄酮葡萄糖苷及芒柄花素)、异黄酮类及其苷类、皂苷类等化合物[12-13]。方中臣药防风含有色原酮、挥发油及多糖类等有效成分,其中色原酮类具有解热、镇痛、抗炎、抗肿瘤等多种功效,故选取升麻素苷、升麻素作为制剂中防风药材的质控指标[14-15]。白术健脾益气,助黄芪以加强益气固表之功效,为佐药,特征成分白术内酯Ⅲ、白术内酯Ⅱ及白术内酯Ⅰ在HPLC图中可清晰分辨[16-17]。

表3 玉屏风丸7 组分加样回收试验结果(n=6)
3.2 色谱条件优化
本研究中对样品提取方法(超声、浸渍过夜、回流)、提取溶剂(甲醇、85%甲醇、65%甲醇、55%甲醇)、提取时间(25,35,45 min)、流动相(甲醇、乙腈作为有机相,水、0.1%磷酸溶液、0.3%磷酸溶液、0.5%磷酸溶液、0.5%冰醋酸作为水相)、柱温(25,30,35 ℃)进行了详细考察,结果显示,85%甲醇作溶剂,超声处理45 min,柱温35 ℃,流动相为乙腈-0.5%磷酸溶液梯度洗脱时基线平稳,保留时间适中,指纹图谱色谱峰数量多且分离效果佳,各色谱峰峰形好。采用DAD 检测器在190 ~400 nm 波长处扫描,为使各成分均在其最大吸收波长下进行测定,采用不同时段切换不同检测波长:0 ~20 min时设定检测波长为300 nm,测定升麻素苷;20 ~40 min时设定检测波长为254 nm,检测毛蕊异黄酮葡萄糖苷、芒柄花素及升麻素;40 ~46 min 时设定检测波长为220 nm,检测白术内酯Ⅱ和白术内酯Ⅲ;46 ~60 min 时设定检测波长为275 nm,检测白术内酯Ⅰ。
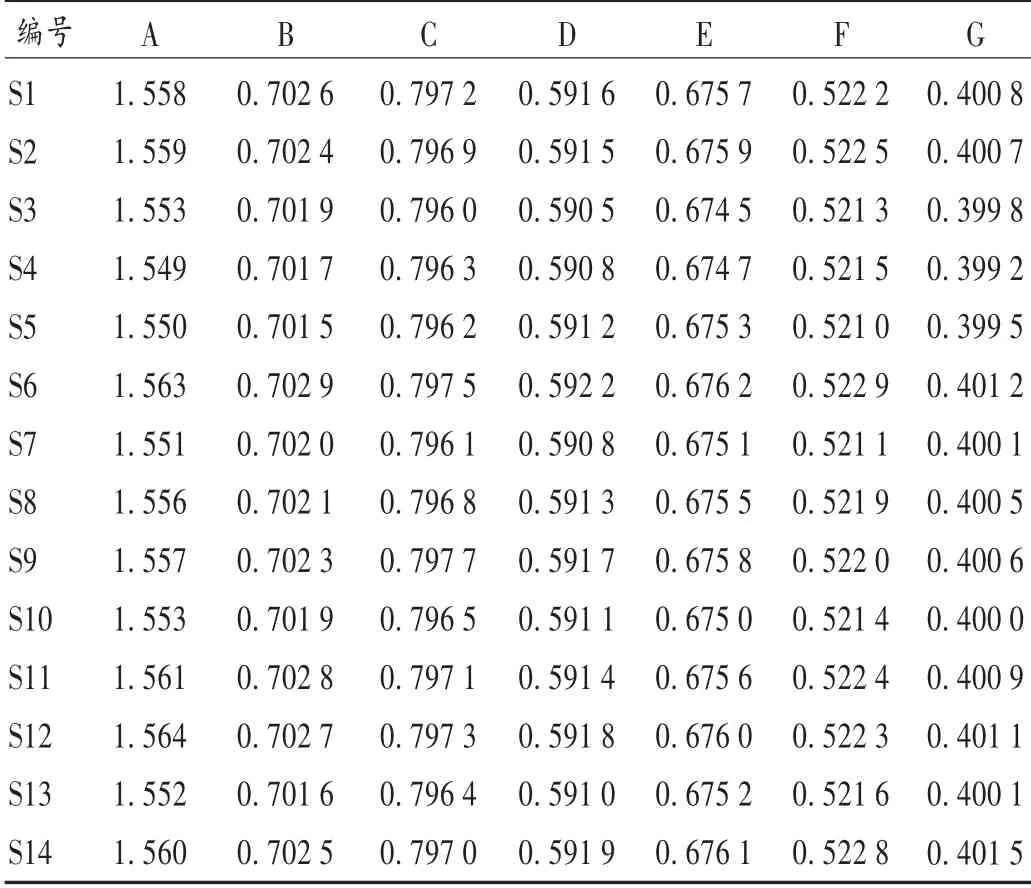
表4 玉屏风丸中7 组分含量测定结果(mg/g,n=3)
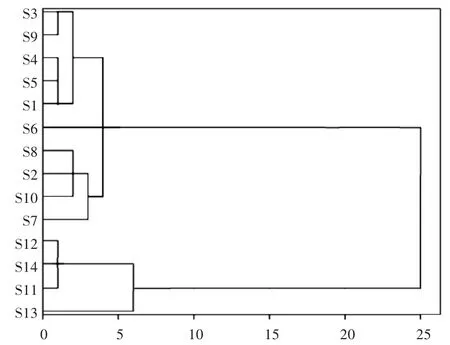
图4 14 批玉屏风丸聚类分析树状图
3.3 方法评价
基于中成药的质量控制的特点,本研究中建立了玉屏风丸指纹图谱,实现了对其多指标成分进行整体综合的客观评价与分析,保证药效的稳定性;同时又建立毛蕊异黄酮葡萄糖苷、芒柄花素、升麻素苷、升麻素、白术内酯Ⅲ、白术内酯Ⅱ、白术内酯Ⅰ的含量测定方法,实现玉屏风丸的定量分析。该方法前处理条件简便、快捷,测定结果准确、重复性好、实用性强,同时具备定性和定量双重评价功能,可有效控制玉屏风丸的质量。
