时间分辨同步荧光法同时检测水中硫酸新霉素和磺胺二甲基嘧啶含量
2020-03-11刘木华袁海超黄双根赵进辉宁王
陈 健 刘木华 袁海超 黄双根 赵进辉 徐 宁王 婷 胡 围
(江西农业大学江西省现代农业装备重点实验室/江西省果蔬采后处理关键技术及质量安全协同创新中心,江西南昌 330045)
硫酸新霉素(neomycin sulfate,NEO)和磺胺二甲基嘧啶(sulfamethazine,SM2)均属于广谱抗生素,对多种致病菌具有抑制或杀灭的效果。它们被广泛应用于水产养殖业、畜牧业和治疗疾病等领域[1-2]。然而,这些抗生素的大量使用必然会给生态环境带来巨大压力,特别是未被生物体有效吸收的药物会直接进入水环境,不仅危害生态系统安全,而且会危害人体健康[3-4]。研究表明,现有的水处理工艺不能彻底去除水体中的痕量抗生素[5-7],我国作为抗生素生产和使用大国,水中抗生素污染比其他国家或地区更加严重[8-9]。因此,迫切需要一种快速、便捷和可靠的水中抗生素检测方法来规范水中抗生素污染的现象。
目前,高效液相色谱法[10-12]、免疫分析法[13-14]、毛细管电泳法[15-16]等均已被用于水中抗生素的检测。其中高效液相色谱法抗干扰能力强、灵敏度高,但是运行成本高,不易推广;免疫分析法样品处理较为困难,部分环节成本高;毛细管电泳法重复性差。而荧光分析法因其具有线性范围大、灵敏度高以及可供选择的参数多等优点,在诸多领域得到了应用。NEO 属于氨基糖苷类抗生素,具有氨基糖结构,SM2 属于磺胺类抗生素,具有芳伯胺基结构,NEO 和SM2 本身不发荧光,但它们与邻苯二甲醛在还原剂2-巯基乙醇的作用下会生成有荧光特性的异吲哚衍生物[17-19],生成路线见图1。
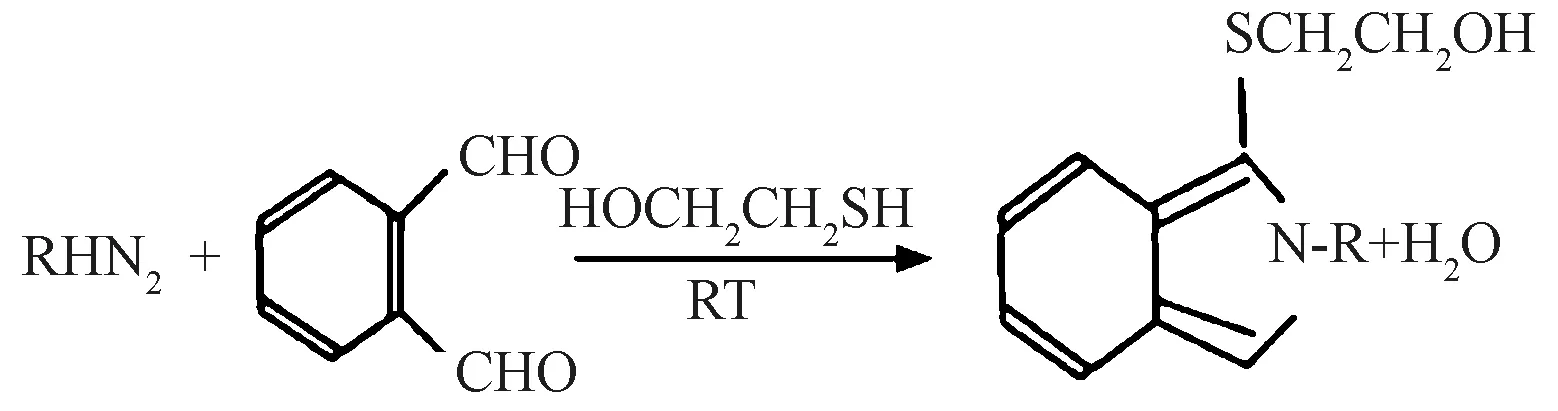
图1 邻苯二甲醛与伯胺类化合物衍生反应示意图Fig.1 Schematic diagram of derivative reaction of o-phthalaldehyde with primary amines
目前关于NEO 和SM2 的荧光法检测方法大多是对禽肉、禽蛋、牛奶等的检测,如徐将等[17]采用荧光法建立了鸭肉中NEO 的测定方法,检测限为1.0 mg·L-1;赵进辉等[20]采用导数同步荧光光谱、小波、分段遗传算法(subsection genetic algorithm,SGA)和最小二乘支持向量回归(least square support vector regression,LSSVR)联用建立了鸭蛋中NEO 的预测模型,模型预测集的决定系数(R2)为0.967 1;邓樱花等[21]采用荧光法建立了蛋清和牛奶中SM2 的检测方法,检出限为2.8 μg·L-1。而有关水中NEO 或SM2 采用荧光法检测的相关报道较少,尤其是水中NEO 和SM2 采用荧光法同时检测,鲜见相关报道。同步荧光法具有光散射少、选择性高和简化图谱等优势[22]。时间分辨荧光法适用于对混合体系中光谱重叠但荧光寿命有差异的组分进行检测[23-24]。时间分辨同步荧光法结合了时间分辨荧光法与同步荧光法的优点,能避免水中NEO、SM2 与邻苯二甲醛衍生物的同步荧光光谱重叠[25]。本研究采用时间分辨同步荧光法同时对它们进行定性、定量解析,尝试建立一种水中NEO 和SM2 含量快速检测的方法。
1 材料与方法
1.1 材料与试剂
NEO 标准品(分析标准品),上海阿拉丁生化科技股份有限公司;SM2 标准品(99.4%),德国Dr.Ehrenstorfer 公司;邻苯二甲醛(99.0%),成都艾科达化学试剂有限公司;2-巯基乙醇(99.0%),成都艾科达化学试剂有限公司;硼酸(≥99.5%),西陇科学股份有限公司;磷酸(≥85.0%),西陇化工股份有限公司;冰乙酸(≥99.5%),天津市大茂化学试剂厂。
1.2 主要仪器与设备
Cary Eclipse 荧光分光光度计,Varian,Inc.美国;FA1004B 型电子天平(精度为0.1 mg),上海上平仪器有限公司;石英比色皿(1 cm 光程),宜兴市晨伟玻璃仪器厂;全自动RO 纯水机,湖南科尔顿水务有限公司;JK-50B 型超声波清洗器,合肥金尼克机械有限公司。
1.3 试验方法
1.3.1 标准液及试剂溶液的配制 100.0 mg·L-1NEO 标准贮备液配制:准确称取NEO 标准品10.0 mg于100.0 mL 容量瓶中,先用少量超纯水溶解,待超声后,冷却至室温,再用超纯水定容,配制成浓度为100.0 mg·L-1的NEO 标准贮备液;100.0 mg·L-1SM2标准贮备液配制:准确称取SM2 标准品10.0 mg 于100.0 mL 容量瓶中,先用少量超纯水溶解,待超声后,冷却至室温,再用超纯水定容,配制成浓度为100.0 mg·L-1的SM2 标准贮备液。8.0 mg·L-1NEO 标准工作液配制:准确量取NEO 标准贮备液8.0 mL 于100.0 mL 容量瓶中,用超纯水稀释至刻度,配制成浓度为8.0 mg·L-1的NEO 标准工作液;8.0 mg·L-1SM2 标准工作液配制:准确量取SM2 标准贮备液8.0 mL 于100.0 mL 容量瓶中,用超纯水稀释至刻度,配制成浓度为8.0 mg·L-1的SM2 标准工作液。不同质量浓度的NEO 与SM2 水溶液配制:分别量取不同体积的100.0 mg·L-1NEO 标准贮备液和100.0 mg·L-1SM2标准贮备液于10.0 mL 容量瓶中,用超纯水定容至10.0 mL,得到不同质量浓度的NEO 与SM2 水溶液(NEO 质量浓度范围为0.5~14.0 mg·L-1,SM2 质量浓度浓度范围为0.25 ~9.0 mg·L-1)。邻苯二甲醛溶液配制:准确称取邻苯二甲醛标准品13.4 mg 于100.0 mL 容量瓶中,先用少量超纯水溶解,待超声后,冷却至室温,再用超纯水定容,配制成浓度为0.001 mol·L-1的邻苯二甲醛溶液;2-巯基乙醇溶液配制:准确量取2-巯基乙醇140 μL 于100.0 mL 容量瓶中,用超纯水稀释至刻度,配制成浓度为0.02 mol·L-1的2-巯基乙醇溶液;BR 缓冲液配制:用超纯水配制浓度各为0.04 mol·L-1的磷酸、乙酸和硼酸混合缓冲溶液。
1.3.2 不同条件下时间分辨同步荧光光谱的采集
1)不同组分的时间分辨同步荧光光谱的采集:①分别取8.0 mg·L-1NEO 水溶液、8.0 mg·L-1SM2 水溶液、8.0 mg·L-1NEO 与SM2 水溶液混合液各2.0 mL 于不同的石英比色皿中。②分别取超纯水8.0 mg·L-1NEO水溶液、8.0 mg·L-1SM2 水溶液、8.0 mg·L-1NEO 与SM2 水溶液混合液各1.0 mL 于不同的石英比色皿中,再先后均加入0.5 mL 0.001 mol·L-1邻苯二甲醛溶液、0.5 mL 0.02 mol·L-12-巯基乙醇溶液和0.5 mL BR 缓冲液(pH 值1.8)。对步骤①、②中的样品,先采集1 min、同步波长差120 nm 下的同步荧光光谱,再采集80min、同步波长差150 nm 下的同步荧光光谱。
2)不同荧光衍生化时间和不同同步波长差时的时间分辨同步荧光光谱的采集:①分别取8.0 mg·L-1NEO 水溶液、8.0 mg·L-1SM2 水溶液、8.0 mg·L-1NEO与SM2 水溶液混合液各1.0 mL 于不同的石英比色皿中,再先后均加入0.5 mL 0.001 mol·L-1邻苯二甲醛溶液、0.5 mL 0.02 mol·L-12-巯基乙醇溶液和0.5 mL BR 缓冲液(pH 值1.8)。②分别取8.0 mg·L-1NEO水溶液、8.0 mg·L-1SM2 水溶液各1.0 mL 于不同的石英比色皿中,再先后均加入0.5 mL 0.001 mol·L-1邻苯二甲醛溶液、0.5 mL 0.02 mol·L-12-巯基乙醇溶液和0.5 mL BR 缓冲液(pH 值1.8)。对步骤①中样品的采集时间均为1 ~120 min,采集时间间隔均为1 min,其中采集120 nm 同步波长差下NEO 水溶液和NEO与SM2 水溶液混合液的同步荧光光谱,采集150 nm同步波长差下SM2 水溶液和NEO 与SM2 水溶液混合物的同步荧光光谱。对步骤②中的样品,分别采集110、115、120、125、130、135、140、145、150、155 和160 nm 同步波长差下的同步荧光光谱,其中NEO 水溶液采集时间1~50 min,SM2 水溶液采集时间1 ~80 min,采集时间间隔均为1 min。
3)不同邻苯二甲醛溶液加入量的时间分辨同步荧光光谱的采集:首先,取8.0 mg·L-1NEO 与SM2 水溶液0.5 mL 于不同的石英比色皿中,再先后分别加入不同体积的0.001 mol·L-1邻苯二甲醛溶液(0.25、0.50、0.75、1.00 和1.25 mL)、0.25 mL 0.02 mol·L-12-巯基乙醇溶液、0.25 mL BR 缓冲液(pH 值1.8)。先采集1 min、同步波长差120 nm 下的同步荧光光谱,再采集80 min、同步波长差150 nm 下的同步荧光光谱。
4)不同2-巯基乙醇溶液加入量的时间分辨同步荧光光谱的采集:首先,取8.0 mg·L-1NEO 与SM2 水溶液0.5 mL 于不同的石英比色皿中,再先后分别加入1.0 mL 0.001 mol·L-1邻苯二甲醛溶液,不同体积的0.02 mol·L-12-巯基乙醇溶液(0.1、0.2、0.25、0.3 和0.4 mL,pH 值1.8)和0.25 mL BR 缓冲液(pH 值1.8)。先采集1 min、同步波长差120 nm 下的同步荧光光谱,再采集80 min、同步波长差150 nm 下的同步荧光光谱。
5)不同BR 缓冲液加入量的时间分辨同步荧光光谱的采集:首先,取8.0 mg·L-1NEO 与SM2 水溶液0.5 mL 于不同的石英比色皿中,再先后分别加入1.0 mL 0.001 mol·L-1邻苯二甲醛溶液、0.25 mL 0.02 mol·L-12-巯基乙醇溶液及不同体积的BR 缓冲液(0.025、0.05、0.1、0.2、0.3 和0.4 mL)。先采集1 min、同步波长差120 nm 下的同步荧光光谱,再采集80 min、同步波长差150 nm 下的同步荧光光谱。
6)定量样品的时间分辨同步荧光光谱的采集:首先,取不同质量浓度的NEO 与SM2 水溶液0.5 mL 于不同的石英比色皿中,再先后加入1.0 mL 0.001 mol·L-1邻苯二甲醛溶液、0.25 mL 0.02 mol·L-12-巯基乙醇溶液和0.025 mL BR 缓冲液。先采集1 min、同步波长差120 nm 下的同步荧光光谱,再采集80 min、同步波长差150 nm 下的同步荧光光谱。
1.4 荧光光谱仪参数设置
使用Cary Eclipse 荧光分光光度计采集时间分辨同步荧光光谱。采集参数如下:同步激发波长扫描范围230~400 nm,PMT 探测器电压为700 Ⅴ,激发和发射狭缝为10 nm,扫描速度为210 nm·min-1,平滑方式为Moving average。每个样品均重复测量5 次。
1.5 数据分析
光谱预处理方法:采用The Unscrambler X 10.4 对试验数据进行处理,将采集的原始荧光光谱数据导入此软件进行基线偏移操作。
同步荧光衰变曲线数据处理:提取1 ~50 min 内NEO 与邻苯二甲醛衍生物335 nm 波长处的同步激发特征峰,以时间(min)为X轴、同步波长差(nm)为Y轴、同步激发特征峰强度(a.u.)为Z轴,在Origin 2018中建立NEO 与邻苯二甲醛衍生物的同步荧光衰变曲线;提取1 ~80 min 内SM2 与邻苯二甲醛衍生物291 nm 波长处的同步激发特征峰,以时间(min)为X轴、同步波长差(nm)为Y轴、同步激发特征峰强度(a.u.)为Z轴,在Origin 2018 中建立SM2 与邻苯二甲醛衍生物的同步荧光衰变曲线。
训练集数据处理:在10 个样本中挑选5 个样本作为训练集样本,以水溶液质量浓度X(mg·L-1)为横坐标、同步激发特征峰的强度Y(a.u.)为纵坐标,在Origin 2018 中绘制水溶液中NEO 和SM2 的工作曲线,并进行线性回归分析。
预测集数据处理:取样本集中剩下的5 个样本作为测试集样本,以真实值(mg·L-1)为横坐标、预测值(mg·L-1)为纵坐标,分别在Origin 2018 中绘制水溶液中NEO 和SM2 的真实值与预测值之间的关系曲线。
2 结果与分析
2.1 时间分辨同步荧光图谱
图2-A、B 分别是采集时间为1 min、同步波长差为120 nm 的时间分辨同步荧光图谱和采集时间为80min,同步波长差为150 nm 的时间分辨同步荧光图谱。在同步波长差为120 nm 时,NEO 衍生体系的同步激发特征峰是位置在335 nm 波长处的宽谱峰;在同步波长差为150 nm 时,SM2 衍生体系的同步激发特征峰是位置在291 nm 波长处的宽谱峰。
在1 min 的时间分辨同步荧光图谱中超纯水体系、NEO 水溶液、SM2 水溶液、NEO 与SM2 水溶液和SM2 衍生体系均无明显的荧光峰,NEO 衍生体系和NEO 与SM2 衍生体系具有335 nm 波长处的荧光峰(图2-A)。
在80 min 的时间分辨同步荧光图谱中超纯水体系、NEO 水溶液、SM2 水溶液和NEO 与SM2 水溶液均无明显的荧光峰,SM2 衍生体系和NEO 与SM2 衍生体系具有291 nm 波长处的荧光峰;NEO 衍生体系的荧光强度远弱于SM2 衍生体系的荧光强度,且两者间的特征峰峰位相差很大(图2-B)。
综合分析,可以应用时间分辨同步荧光法测定NEO 与SM2 衍生体系中的NEO 和SM2 含量。
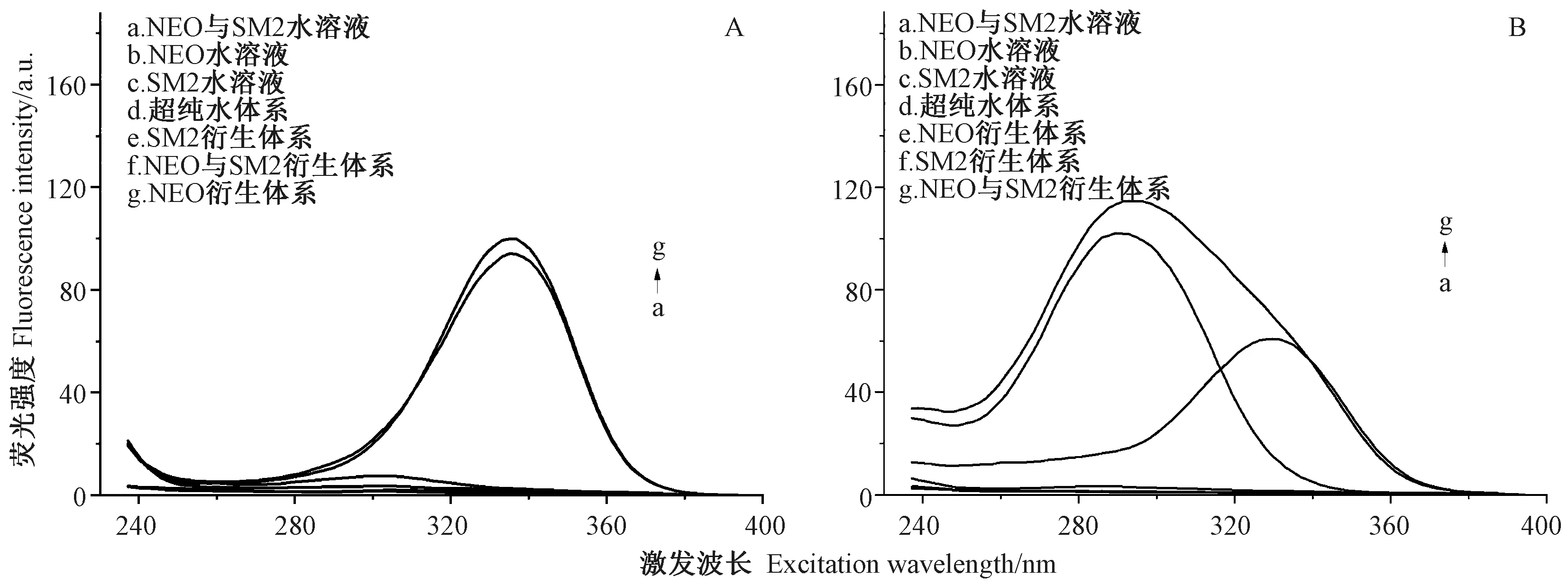
图2 1 min 时间分辨同步荧光图谱(A)与80 min 时间分辨同步荧光图谱(B)Fig.2 Time-resolved synchronous fluorescence spectra at 1 min(A)and 80 min(B)
2.2 荧光衍生化时间及同步波长差对水中NEO 和SM2 的同步荧光强度的影响
NEO、SM2 与邻苯二甲醛反应生成异吲哚衍生物。显然,荧光衍生化时间会影响异吲哚衍生物的生成量。在同步扫描过程中,同步波长差的选择将直接影响同步荧光光谱的信号强度。由图3 可知,1 ~50 min 内随着荧光衍生化时间的延长,NEO 与邻苯二甲醛衍生物的同步荧光强度呈快速减弱的趋势。由图4-A 可知,随着同步波长差的增加,NEO 与邻苯二甲醛衍生物的同步荧光强度呈先波动增强后波动减弱的趋势,在同步波长差120 nm 时最强。
由图3 可知,在1 ~80 min 内SM2 与邻苯二甲醛衍生物的同步荧光强度变化趋势与NEO 与邻苯二甲醛衍生物的趋势相反,呈快速增强的趋势,在80 min时最强。由图4-B 可知,随着同步波长差增加,SM2 与邻苯二甲醛衍生物的同步荧光强度与NEO 与邻苯二甲醛衍生物的趋势相同,呈先波动增强后波动减弱的趋势,在同步波长差150 nm 时最强。
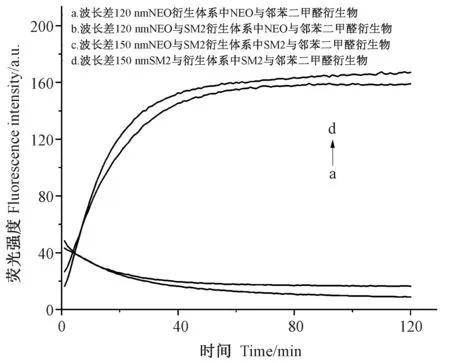
图3 NEO 衍生体系和SM2 衍生体系分别在120 和150 nm 波长差下的同步荧光衰变曲线以及NEO 与SM2 衍生 体系在120 和150 nm 波长差下同步荧光衰变曲线Fig.3 Synchronous fluorescence decay curves of NEOderived and SM2-derived systems at wavelength differences of 120 and 150 nm,respectively,and synchronous fluorescence decay curves of NEO and SM2 derived systems at wavelength differences of 120 and 150 nm
综合分析,NEO 与邻苯二甲醛衍生物和SM2 与邻苯二甲醛衍生物在合适的荧光衍生化时间均具有很强的荧光信号,而它们的荧光衰变趋势相反,可以利用这个特性在时间上对两种物质进行分辨。本研究选择NEO 采集时间1 min、同步波长差120 nm;SM2 选择采集时间80 min、同步波长差150 nm,进行后续试验。
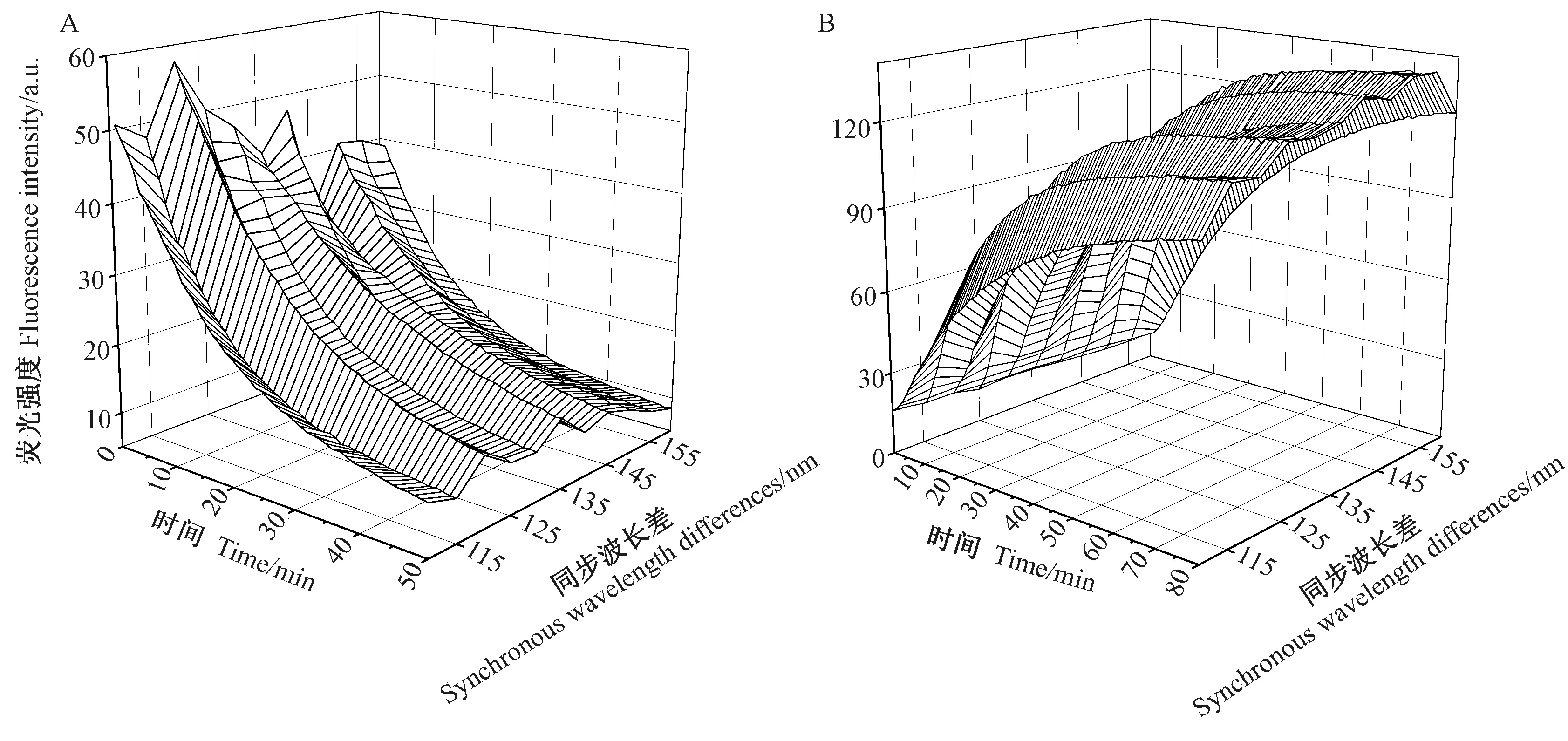
图4 不同同步波长差下NEO 衍生体系同步荧光衰变曲线(A)与SM2 衍生体系同步荧光衰变曲线(B)Fig.4 Synchronous fluorescence decay curve of NEO-derived system(A)as well as SM2-derived system(B)under different synchronous wavelength differences
2.3 邻苯二甲醛溶液加入量对水中NEO 和SM2 的同步荧光强度的影响
邻苯二甲醛是常用的荧光衍生试剂,它在强还原剂存在的条件下会与NEO 和SM2 的反应生成异吲哚衍生物[26]。因此,为提高水中NEO 和SM2 的荧光信号,需要分析邻苯二甲醛溶液加入量以获得较优的荧光信号。由图5 可知,随着邻苯二甲醛溶液加入量的增加,NEO 与邻苯二甲醛衍生物的同步荧光强度呈先增强后减弱的趋势,在邻苯二甲醛溶液加入量为1.0 mL 时,NEO 与邻苯二甲醛衍生物的同步荧光强度最佳,但稍弱于SM2 与邻苯二甲醛衍生物的同步荧光强度。随着邻苯二甲醛溶液加入量的增加,SM2 与邻苯二甲醛衍生物的同步荧光强度则呈快速减弱的趋势,在邻苯二甲醛溶液加入量为0.25 mL 时,SM2 与邻苯二甲醛衍生物的同步荧光强度最佳,但此时NEO 与邻苯二甲醛衍生物的同步荧光强度较弱。在分析单一组分时,选择邻苯二甲醛溶液加入量的主要依据是获得该组分的最佳荧光强度;但在同时分析两组分时,不仅要考虑各组分单一的荧光强度,还需要考虑各组分同时的荧光强度。综合分析,在邻苯二甲醛溶液加入量为1.0 mL 时,NEO 与邻苯二甲醛衍生物的同步荧光强度最佳,且SM2 与邻苯二甲醛衍生物的同步荧光强度强于NEO 与邻苯二甲醛衍生物,因此,本研究选择邻苯二甲醛溶液加入量为1.0 mL。
有报道称,NEO 与邻苯二甲醛在碱性环境下反应更加充分,其最适宜反应的pH 值在10.5 左右[27]。而SM2 与邻苯二甲醛在强酸性环境下反应更加充分,其最适宜反应的pH 值在1.5 左右[21]。因此,体系的pH值升高会促进NEO 与邻苯二甲醛反应,但会抑制SM2与邻苯二甲醛反应。综合考虑,本研究用BR 缓冲液将体系调至偏酸性。在室温下,邻苯二甲醛溶液呈中性,加入到体系中,体系的pH 值会逐渐升高接近中性。
邻苯二甲醛溶液加入量为0.25 ~1.0 mL 时,随着邻苯二甲醛溶液加入量增加,体系的邻苯二甲醛含量及pH 值均升高,共同促进NEO 与邻苯二甲醛的反应。因此,NEO 与邻苯二甲醛衍生物的同步荧光强度呈上升趋势;邻苯二甲醛溶液加入量为1.0 ~1.75 mL时,体系的pH 值变化趋于稳定,同时NEO 与邻苯二甲醛基本得到充分反应,此时邻苯二甲醛溶液的增加会降低NEO 与邻苯二甲醛衍生物的浓度,进而减弱了NEO 与邻苯二甲醛衍生物的同步荧光强度。
此外,邻苯二甲醛溶液加入量为0.25 ~1.75 mL时,随着邻苯二甲醛溶液加入量增加,体系邻苯二甲醛含量升高,促进了SM2 与邻苯二甲醛反应,但是体系pH 值升高,抑制了SM2 与邻苯二甲醛反应(图5),SM2 与邻苯二甲醛衍生物的同步荧光强度呈下降趋势,此时抑制作用占据主导地位。
2.4 2-巯基乙醇溶液加入量对水中NEO 和SM2 的同步荧光强度的影响
本研究所涉及的荧光衍生化反应是以邻苯二甲醛为衍生剂,2-巯基乙醇为还原剂,NEO 和SM2 与邻苯二甲醛反应生成具有荧光特性的异吲哚衍生产物[20]。因此,优化2-巯基乙醇溶液加入量有助于增强NEO和SM2 水溶液衍生体系的荧光信号。由图6 可知,随着2-巯基乙醇溶液加入量的增加,NEO 与邻苯二甲醛衍生物的同步荧光强度呈先增强后减弱的趋势,在2-巯基乙醇溶液加入量为0.2 mL 时,NEO 与邻苯二甲醛衍生物的同步荧光强度最佳,此时SM2 与邻苯二甲醛衍生物的同步荧光强度约是其四分之三;随着2-巯基乙醇溶液加入量增加,SM2 与邻苯二甲醛衍生物的同步荧光强度呈快速增强趋势,在邻苯二甲醛溶液加入量为0.4 mL 时,SM2 与邻苯二甲醛衍生物的同步荧光强度最佳,但此时NEO 与邻苯二甲醛衍生物的同步荧光强度较弱。在2-巯基乙醇溶液加入量为0.25 ~0.3 mL 时,二者的同步荧光强度曲线相交,此时这两种衍生物的同步荧光强度相差不大。综合分析,在2-巯基乙醇溶液加入量为0.25~0.3 mL 时,这两种衍生物的同步荧光强度相近;又因同步荧光强度曲线交点更靠近0.25 mL,所以本研究选择2-巯基乙醇溶液加入量为0.25 mL。
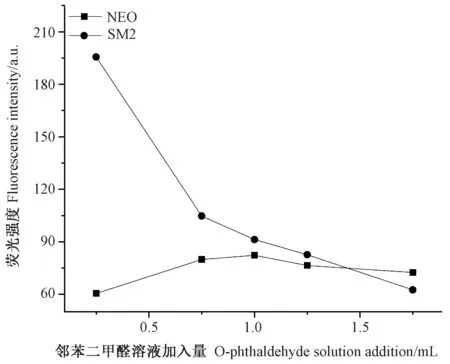
图5 不同邻苯二甲醛溶液加入量对同步荧光强度的影响Fig.5 Effect of different o-phthaldehyde solution additions on synchronous fluorescence intensity
在室温下,2-巯基乙醇溶液呈酸性,pH 值介于4.5~6.0 之间。2-巯基乙醇溶液加入到体系中,体系的pH 值降低,进而抑制NEO 与邻苯二甲醛反应,但会促进SM2 与邻苯二甲醛反应。
2-巯基乙醇溶液加入量为0.1~0.2 mL 时,随着2-巯基乙醇溶液加入量的增加,体系的2-巯基乙醇含量升高,促进NEO 与邻苯二甲醛的反应,但同时体系pH 值降低,抑制了NEO 与邻苯二甲醛的反应(图6),NEO 与邻苯二甲醛衍生物的同步荧光强度呈上升趋势,此时促进作用占据主导地位;2-巯基乙醇溶液加入量为0.2~0.4 mL 时,随着2-巯基乙醇溶液加入量的增加,体系pH 值持续降低引起NEO 与邻苯二甲醛衍生物的同步荧光强度呈下降趋势,此时抑制作用占主导地位。
此外,2-巯基乙醇溶液加入量为0.1 ~0.4 mL 时,随着2-巯基乙醇溶液加入量的增加,体系的2-巯基乙醇含量升高及pH 值降低,共同促进了SM2 与邻苯二甲醛的反应,因此,SM2 与邻苯二甲醛衍生物的同步荧光强度呈上升趋势。
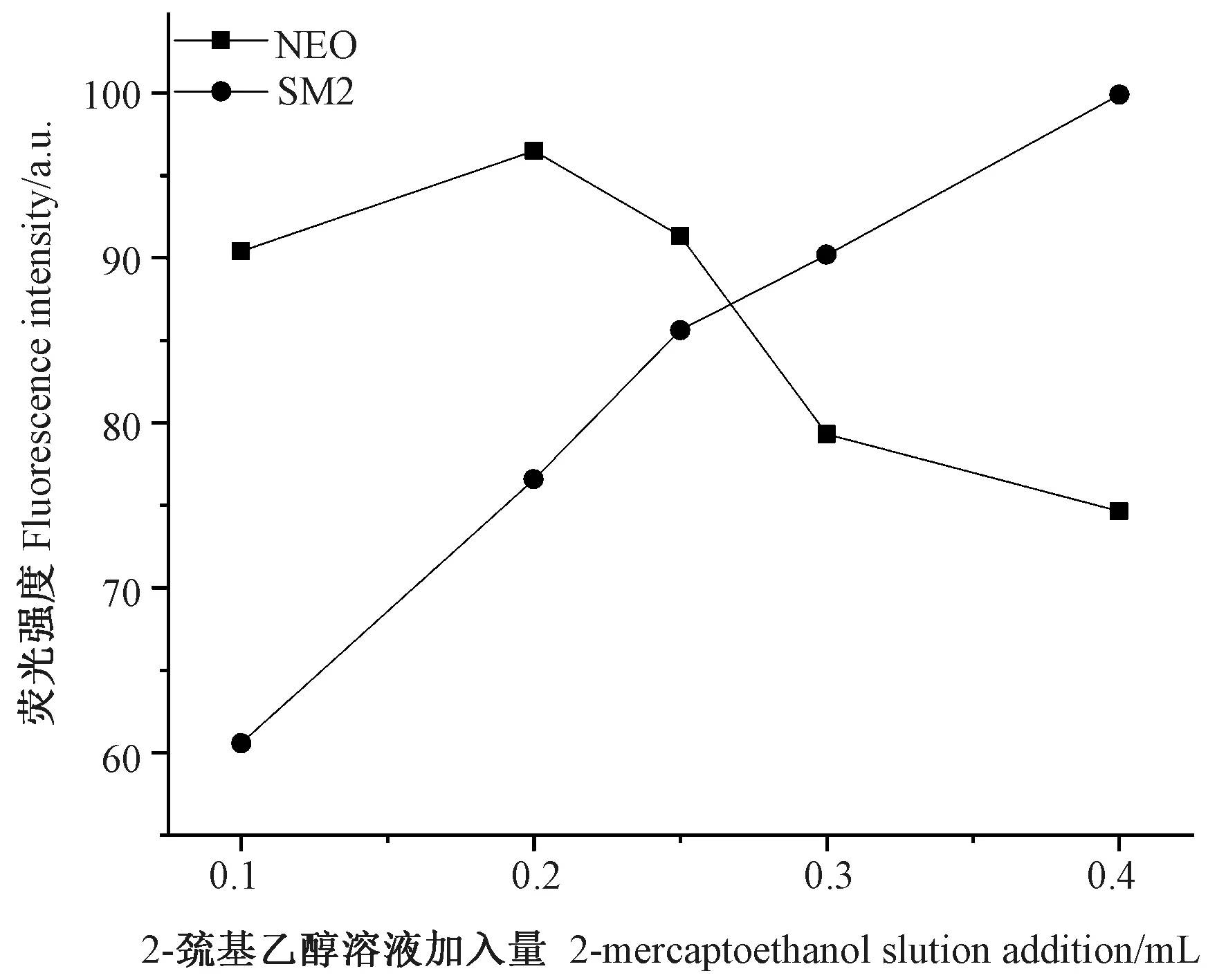
图6 不同2-巯基乙醇溶液加入量对同步荧光强度的影响Fig.6 Effect of different 2-mercaptoethanol solution additions on synchronous fluorescence intensity
2.5 BR 缓冲液加入量对水中NEO 和SM2 的同步荧光强度的影响
溶液pH 值会在很大程度上影响荧光强度[28],因此,优化BR 缓冲液加入量有助于增强NEO 和SM2 水溶液衍生体系的荧光信号。由图7 可知,随着BR 缓冲液加入量的增加,NEO 与邻苯二甲醛衍生物和SM2与邻苯二甲醛衍生物的同步荧光强度均呈减弱趋势。因此,综合考虑同步荧光光谱情况,本研究选择BR 缓冲液加入量为0.025 mL。
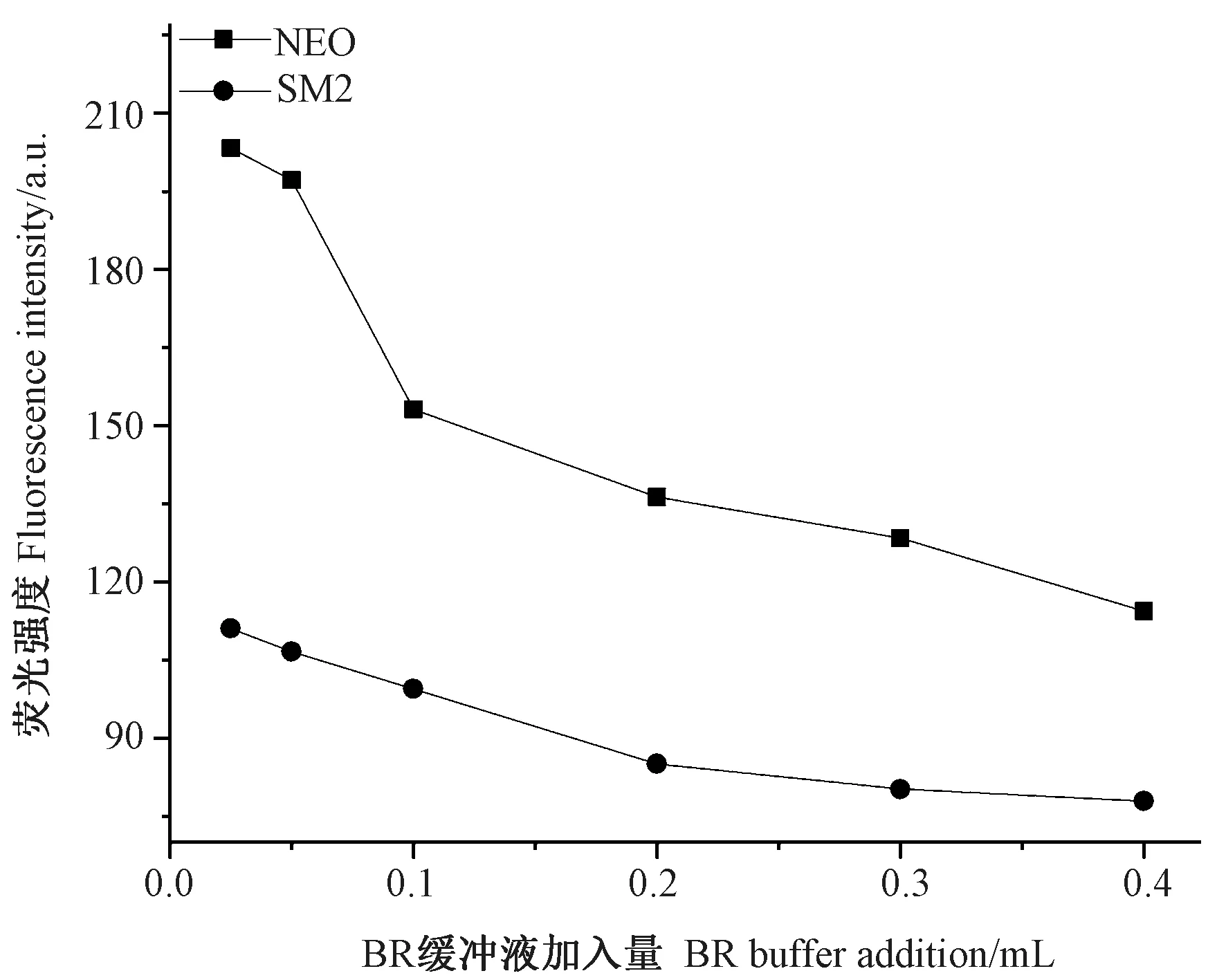
图7 不同BR 缓冲液加入量对同步荧光强度的影响Fig.7 Effect of different BR buffer additions on synchronous fluorescence intensity
2.6 模型的预测与分析
分析采集的含有不同NEO 和SM2 质量浓度的样本荧光光谱发现,随着NEO 和SM2 质量浓度的增加,两者的同步荧光强度随之增强。因此,以同步激发特征峰强度为依据分别对水中NEO 和SM2 的含量进行定量分析。
分析含不同NEO 质量浓度为0.5 ~14.0 mg·L-1的样本荧光光谱强度,不同NEO 浓度与其对应浓度在335 nm 波长处同步激发特征峰强度的关系曲线如图8-A 所示,呈良好的线性关系,线性方程为Y=14.73X+6.14,训练集决定系数(R2C)为0.997 5,检出限为0.5 mg·L-1。为验证研究的可靠性并进一步预测分析,对含不同NEO 浓度(2.0、3.0、6.0、7.0 和13.0 mg·L-1)的样本进行回收试验,由图8-C 可知,预测集样本中NEO 含量的真实值与预测值之间的的决定系数(R2P)为0.998 2,均方根误差(root mean square error of prediction,RMSEP)为0.380 3 mg·L-1。对预测样本进行回收试验,样本回收率处于101.8%~114.0%之间,相对标准偏差(relative standard deviation,RSD)为4.0%~8.4%(n=5)(表1)。
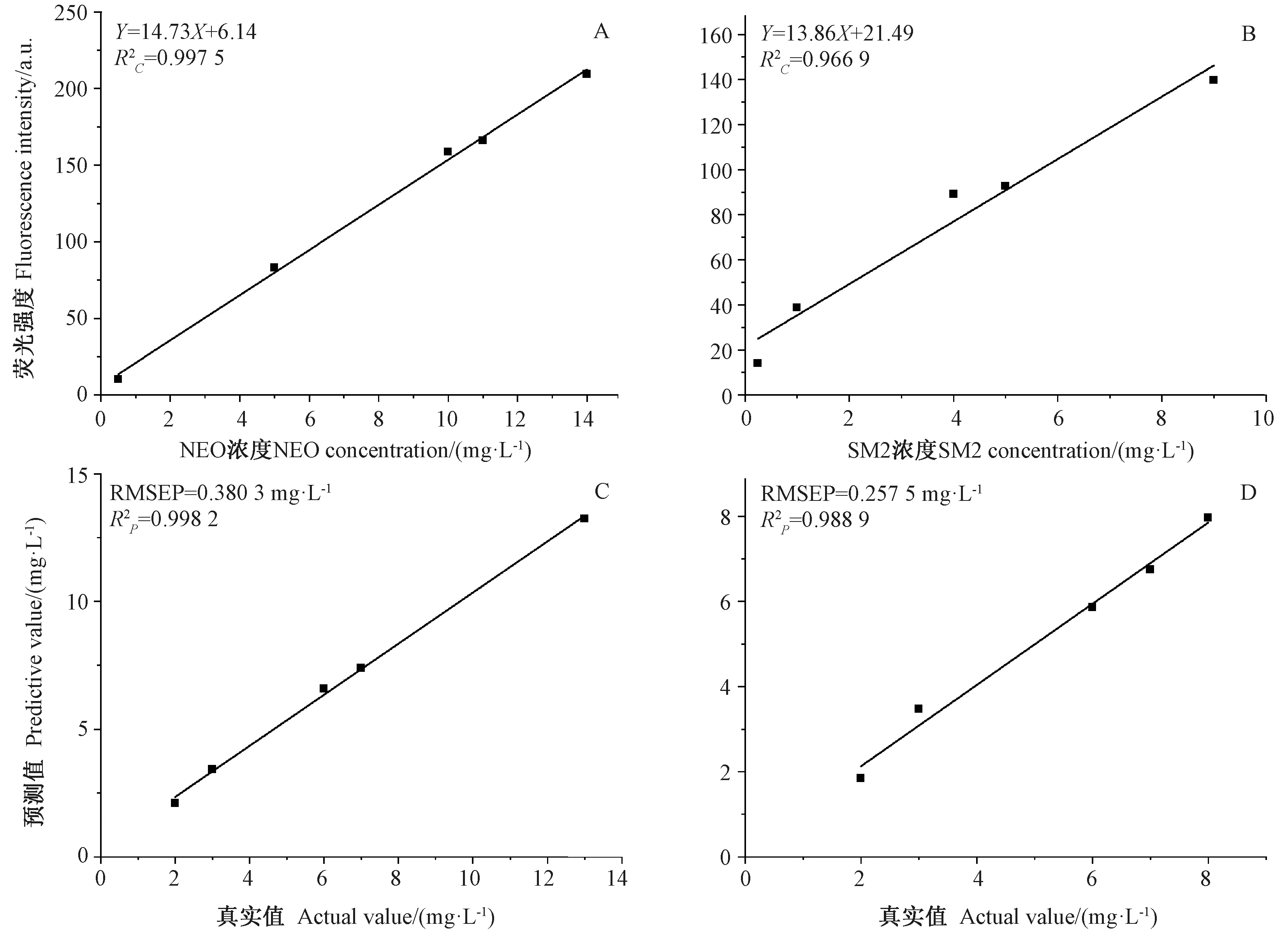
图8 水中NEO(A、C)和SM2(B、D)的工作曲线和预测集样本关系图Fig.8 Working curves aqueous solutions and sample relation of prediction set diagrams of NEO(A,C)and SM2(B,D)
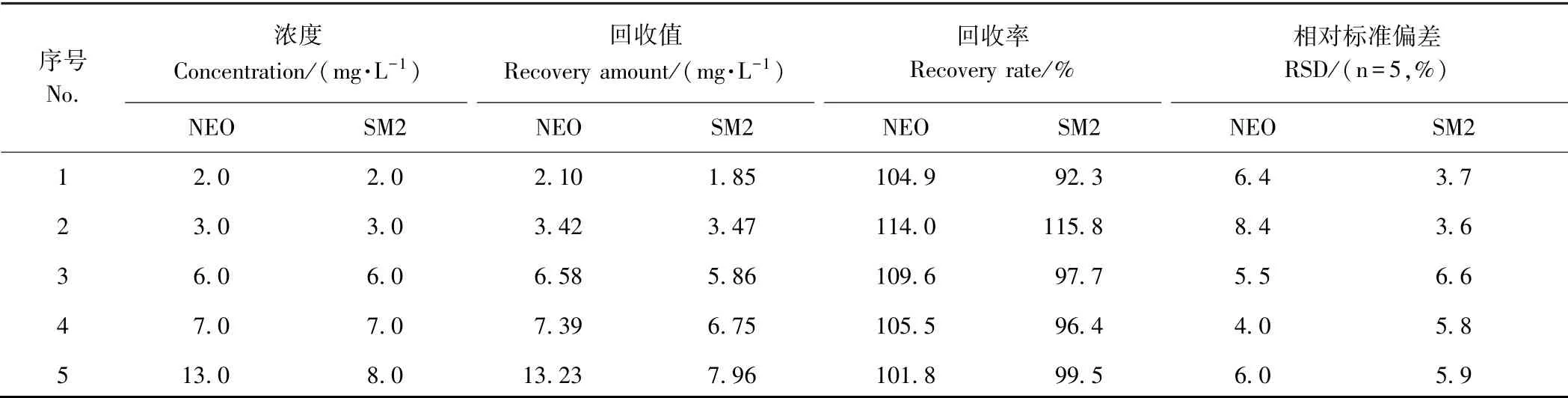
表1 水中NEO 和SM2 的回收率与精密度Table 1 Recovery rate and precision degree of neomycin and sulfadimidine in water
分析含不同SM2 质量浓度为0.25~9.0 mg·L-1的样本的荧光光谱强度,不同SM2 浓度与其对应浓度在291 nm 波长处同步激发特征峰强度的关系曲线(图8-B),呈良好的线性关系,线性方程为Y= 13.86X+21.49,R2C为0.966 9,检出限为0.25 mg·L-1。对含不同SM2 浓度(2.0、3.0、6.0、7.0 和8.0 mg·L-1)的样本进行回收试验。由图8-D 可知,预测集样本中SM2 含量的真实值与预测值之间的R2P为0.988 9,RMSEP 为0.257 5 mg·L-1。对预测样本进行回收试验,样本回收率处于92.3%~115.8%之间,RSD 为3.6%~6.6%(n=5)(表1)。
由检测结果可知,基于本研究的时间分辨同步荧光法应用于同时检测水中NEO 和SM2 含量,能得到较好的准确度和精密度。
3 讨论
随着我国抗生素生产和使用量的持续增长,水中抗生素的污染越来越严重。目前,国内外研究水中NEO 和SM2 含量检测的方法主要采用液相色谱法,如王欣梅等[29]以液相色谱和串联质谱法检测了环境水样中SM2 的含量,检出限为0.06 ng·L-1;Miao 等[30]以微内径柱高效液相色谱和串联质谱法检测了污水处理厂出厂水中NEO 含量,检出限为2.0 pg·L-1。虽然本检测方法的检出限高于液相色谱法的检出限,但本研究所采用的方法操作更加方便、快捷,且设备体积较小,具有现场检测的发展潜力。
由于各组分荧光光谱常会产生重叠,普通荧光法在混合体系中的检测应用难以用于定量分析。与普通荧光法相比,同步荧光法在分析一些光谱重叠、难以分辨的复杂混合物中更具有优势。目前,同步荧光分析在混合体系荧光分析中已有较多应用。如庄宇等[31]提出了基于同步荧光结合神经网络同时测定3 种抗生素(乳酸环丙沙星、乳酸左氧氟沙星、盐酸左氧氟沙星)的方法;张晶玉等[32]进行了基于同步、导数和卡尔曼滤波荧光法的罗丹明B 和罗丹明6G 同时测定的比较研究,发现应用同步荧光法效果最佳。此外,对于光谱重叠但荧光寿命有差异的混合体系还可以采用时间分辨荧光法进行检测。如Leivo 等[33]以时间分辨荧光和荧光免疫法检测了牛奶中氟喹诺酮含量;张笑河等[34]提出了一种时间分辨荧光法同时测定混合液(己酸乙酯和乙酸乙酯)中各组分浓度的方法。本研究结合同步荧光法和时间分辨荧光法,针对水中NEO 和SM2 的含量建立了时间分辨同步荧光检测方法,为荧光分析法实现水中NEO 和SM2 含量的快速、在线和精确检测提供了一定的借鉴。荧光分析法在检测水中NEO 和SM2 含量中的应用仍存在一定的局限性,本研究所能达到的灵敏度、准确度有限,还需要对试验进行完善和优化,有待进一步深入研究。
4 结论
利用同步荧光光谱法结合时间分辨、化学计量学等方法,建立了线性模型预测水中NEO 和SM2 的含量,并对同步波长差、荧光衍生化时间、邻苯二甲醛溶液加入量、2-巯基乙醇溶液加入量和BR 缓冲液加入量进行了优化,结果得到水中不同浓度的NEO 回收率101.8%~114.0%,RSD 为4.0%~8.4%;水中不同浓度的SM2 回收率为92.3%~115.8%,RSD 为3.6%~6.6%。表明采用时间分辨同步荧光法能可有效分辨NEO 和SM2 的邻苯二甲醛衍生物之间的同步荧光光谱,可有效筛选出同步激发特征峰,使水中NEO 和SM2 含量同时被检测,同时简化了检测步骤,为水中NEO 和SM2 含量同时快速检测提供了一种新的方法。
