基于高通量测序的麦红吸浆虫转录组分析
2020-03-09蒋月丽武予清巩中军刘启航
蒋月丽,李 彤,武予清,苗 进,巩中军,段 云,刘启航
(河南省农业科学院植物保护研究所,河南省农作物病虫害防治重点实验室,农业部华北南部有害生物治理重点实验室,郑州 450002)
麦红吸浆虫Sitodiplosismosellana(Gehin)属双翅目瘿蚊科Cecidomyiidae,是一种世界性的小麦害虫,主要分布在美国、加拿大、欧洲、俄罗斯和中国(Lambetal., 2000; Berzonskyetal., 2003),以幼虫潜伏在颖壳内吸食正在灌浆的汁液,造成麦粒瘪疮、空壳或霉烂而减产,具有很大的危害性,一般减产10%~20%,重者减产30%~50%,严重的乃至颗粒无收,全球每年因麦红吸浆虫的危害而造成的经济损失高达数亿美元(Doane and Olfert, 2008; Oakley, 2008)。由于麦红吸浆虫具有虫体小,在土壤中滞育时间长,在田间呈岛屿型分布和发生危害具有隐蔽性、间歇性和周期性的特点(Wise and Lamb, 2004; Liatukasetal., 2009; 郁振兴等,2011),长期以来一直是威胁小麦生产的重要害虫。目前,关于吸浆虫的研究大多集中在生态和生物化学(Hinks and Doane, 1988; Hu and Zhang, 1995; Wise and Lamb, 2004; Wu and Yuan, 2004; Chenetal., 2009),但是由于基因组和转录组信息的缺乏,很少有关于其功能基因的研究,本文通过获得大量转录组信息,可以为其功能基因的挖掘提供巨大的信息资源。
转录组(transcriptome)是指细胞在特定状态下全部表达的RNA的总和,反映相同基因在不同条件下表达水平的差异,并能揭示不同基因的相互作用及各自功能(Velculescu, 1997)。转录组学是一门基于转录组在整体水平之上研究生物基因转录情况的学科。近年来,转录组学技术在揭示细胞生理活动规律和代谢机理的研究中广泛应用(Lockhart & Winzeler, 2008)。转录组测序(RNA sequencing)分为三代。第一代测序技术以基因表达系列分析技术(Serial Analysis of GeneExpression,SAGE)和大规模平行测序技术(Massively Paralel Signature Sequencing,MPSS)为代表,以双脱氧终止法(Sangeretal., 1997)和化学降解法(Whitfeldetal., 1954; Maxametal., 1977)为基础。第二代测序技术—高通量测序技术(High-throughput sequencing)主要包括Roche 454焦磷酸测序技术、Illumina/Solexa测序技术、Solexa测序技术和SOLiD测序技术等,其原理为提取样品总RNA后,用高通量测序平台进行样品文库的深度测序,经图像识别,获得DNA序列(张春兰,2012)。第三代测序技术包括Helicos单分子测序仪、Pacific Bioscience的SMRT技术以及Oxford Nanopore Technologies公司的纳米孔单分子测序技术。其中,Illumina测序技术成本低、周期短、通量高,具有明显的优势。转录组测序可用于研究基因功能、可变剪接、新转录本预测和结构性变异(Alagnaetal., 2009; Barakatetal., 2009; Dassanayakeetal., 2010; Lietal., 2010)。相对于传统的芯片杂交平台,转录组测序可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量及更广泛的检测范围。因此,对于缺乏基因组信息的物种而言,采用转录组测序技术可获得大量的转录本信息,从中发掘重要功能基因的重要研究手段(Franssenetal., 2011; 杨楠等,2012;Lietal., 2013)。
本研究中将Illumina HiSeq 2000高通量测序技术应用到麦红吸浆虫转录组研究,将测序得到的大量数据进行拼接与组装,结合生物信息学方法对所获得的Unigene进行基因功能注释、功能分类、代谢途径分析等,研究结果和分析为进一步的分子标记开发和基因功能研究奠定基础,也为麦红吸浆虫的基因组学研究提供丰富的资源。
1 材料与方法
1.1 试虫
麦红吸浆虫土样,与2014年在河南省新乡市辉县采集,将所采集的吸浆虫土样放入春化室进行春化处理,与次年春季移入气候室,待成虫羽化当天收集,选择健壮活虫经处理后,立即放入液氮保存。
1.2 方法
将液氮冷冻保存的试虫送至北京诺禾致源生物信息科技有限公司。使用试剂盒提取RNA。采用琼脂糖凝胶电泳分析RNA降解程度以及是否有污染,Nanodrop检测RNA的纯度(OD260/OD280比值),Qubit对RNA浓度进行精确定量,Agilent 2100精确检测RNA的完整性。将满足测序要求的RNA样本进行转录组测序及原始数据分析。
1.2.1建库流程
样品检测合格后,用带有Oligo (dT)的磁珠富集真核生物mRNA(若为原核生物,则通过试剂盒去除rRNA来富集mRNA)。随后加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,然后加入缓冲液、dNTPs(dNTP中的dTTP用dUTP取代)和DNA polymerase I和RNase H合成二链cDNA,再用AMPure XP beads纯化双链cDNA,之后用USER酶降解含有U的cDNA第二链。纯化的双链cDNA先进行末端修复、加A尾并连接测序接头,再用AMPure XP beads进行片段大小选择。最后进行PCR扩增,并用AMPure XP beads纯化PCR产物,得到最终的文库。文库构建完成后,先使用Qubit 2.0进行初步定量,稀释文库至1.5 ng/μL,随后使用Agilent 2100对文库的insert size进行检测,insert size符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2 nM),以保证文库质量。库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行Illumina HiSeq测序。
1.2.2转录组信息分析
1.2.2.1 测序数据的预处理
测序得到的原始测序序列(Sequenced Reads)或者raw reads,里面含有带接头的、低质量的reads,为了保证信息分析质量,必须对raw reads过滤,得到clean reads,后续分析都基于clean reads。数据处理的步骤如下:(1)去除带接头(adapter)的reads;(2)去除N(N表示无法确定碱基信息)的比例大于10%的reads;(3)去除低质量reads(质量值Qphred <= 20的碱基数占整个reads的50%以上的reads)。
1.2.2.2 Trinity拼接
利用Trinity软件对样品数据进行组装,通过序列之间的overlap信息组装得到重叠群(Contig),然后在局部进行组装得到转录本(Transcripts),最后从局部中挑选最主要的转录本作为单基因簇(Unigene)(Grabherretal., 2011)。
1.2.2.3 转录本的功能注释
拼接获得转录本后,与7个公共数据库(Nr,Nt,Pfam,KOG/COG,Swiss-Prot,KEGG和GO)。中的蛋白质序列进行比对,取阈值e ≤10-10,通过序列相似性进行功能注释。序列相似性比较使用BLAST(Altschuletal.)算法,然后用WEGO软件对所有的Unigene进行GO功能分类统计(Conesaetal., 2005; Yeetal., 2006)。然后对Unigene分别进行蛋白质直系同源数据库(euKaryotic Ortholog Groups,KOG)功能分类和东京基因与基金组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)代谢途径分析(Romanetal., 2003; Minoruetal., 2004)。
1.2.2.4 CDS预测
CDS预测分为两步:1. 将Unigenes按照NR蛋白库,Swissprot蛋白库的优先级顺序进行比对,若比对上,则从比对结果中提取出转录本的ORF编码框信息,并按照标准密码子表将编码区序列翻译成氨基酸序列(按照5′>3′的顺序);2. 对于没有比对上NR和Swissprot蛋白库的序列,或者比对上未预测出结果的序列,则采用estscan (3.0.3)软件预测其ORF,从而得到这部分基因编码的核酸序列和氨基酸序列。
1.2.2.5 SSR分析
利用MISA软件对麦红吸浆虫转录组中筛选得到1 kb以上的Unigene进行简单重复序列(Simple sequence repeats,SSR)位点分析,搜索标准为:单、二、三、四、五、六核苷酸基序(motif)至少重复次数分别为10、8、5、4、3、3,对查找的SSR类型进行特征分析。
2 结果与分析
2.1 麦红吸浆虫转录组数据的组装
转录组样本数据量为27.88 G,总的CG含量平均值为40.26%,数据中96.70%的序列的质量参数Q≥20,93.53%的Q≥30。经过分析,共获得59 257个Unigenes,总长度49 861 164 bp,最短201 bp,最长29 282 bp,平均长度841 bp(图1)。
2.2 Unigene的功能注释、分类和代谢途径分析
2.2.1Unigene在各数据库中的比对
将获得的59 257个Unigenes分别在7个公共数据库中进行比对,取阈值e≤10-10,分别获得18 596、2 868、9 110、14 680、18 912、19 584和11 279个结果,共计95 029个结果。
2.2.2Unigene的GO分类注释
共有19 584个Unigenes在GO数据库中3大类50个功能组中找到对应(图2)。其中生物学进程组中较大的是细胞进程(Cellular process)及新陈代谢进程(Metabolic process);细胞成分组中较大的细胞(Cell)和细胞成分(Cell part);分子功能组中较大的是结合活性(binging)和催化活性(Catalytic activity)。
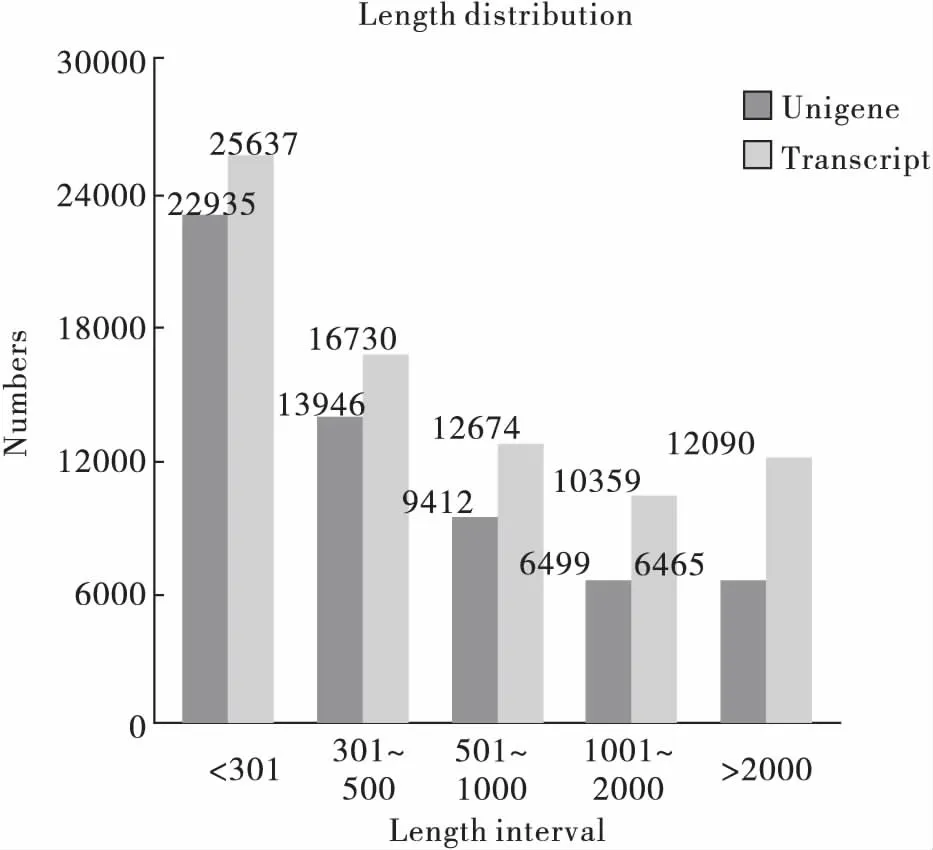
图1 拼接Unigenes与转录本长度分布图Fig.1 length distribution of splice Unigenes and transcript

图2 Unigenes的GO分类图Fig.2 GO functional categories of unigenes
2.2.3Unigene的KOG分类注释
蛋白质直系同源数据库(euKaryotic Ortholog Groups, KOG)是对基因产物进行直系同源分类的数据库。将麦红吸浆虫Unigenes与KOG数据库进行比对,预测Unigenes功能并进行分类统计。研究结果表明,共有11 279个麦红吸浆虫Unigenes被注释,根据其功能大致可分为26类,对每类的Unigenes进行了统计分析(图3 A-Z)。从图中可以看出,Unigenes涉及的KOG功能类别比较全面,涉及了大多数的生命活动。其中,一般功能预测类基因最多(2 226个);其次是信号传导机制类基因(1 467个)、翻译后修饰,蛋白质折叠和分子伴侣(1 265个)、转录(734个);未知蛋白质(1个)最少,其他类别的基因表达丰度都各不相同。

图3 麦红吸浆虫Unigenes的KOG分类图Fig.3 KOG functional categories of Sitodiplosis mosellana Unigenes
2.2.3Unigenes的KEGG分类注释
KEGG(Kyoto Encyclopedia of Genes and Genomes)系统分析基因产物和化合物在细胞中的代谢途径以及这些基因产物的功能的数据库。它整合了基因组、化学分子和生化系统等方面的数据,包括代谢通路(KEGG PATHWAY)、药物(KEGG DRUG)、疾病(KEGG DISEASE)、功能模型(KEGG MODULE)、基因序列(KEGG GENES)及基因组(KEGG GENOME)等等。结合KEGG数据库,对麦红吸浆虫的Unigene可能参与或涉及的代谢途径进行了统计分析。结果显示,共有9 110个麦红吸浆虫Unigenes被注释,分别归属于细胞进程、环境信息进程、遗传信息进程、新陈代谢和有机体系统五大类代谢途径,主要包括细胞生长与死亡、细胞运动、信号转导、能量代谢等32类代谢途径。其中占比例最高的是信号转导有1 078个(占11.83%),其次是转录有730个(占8.01%)和折叠、排序与退化有661个(占7.26%),比例最小的是生物合成与次生代谢仅有52个(占0.57%)(图4)。
2.3 CDS预测
CDS预测结果显示,共30 088条序列可被编码,占全部基因的50.78%(30088/59257)。图5表示所预测CDS的长度统计。
2.4 微卫星位点(SSR)分析
SSR,简单重复序列(simple sequence repeats),又称为短串联重复序列和微卫星标记。是一类由几个核苷酸(1~5个)为重复单位组成的长达几十个核苷酸的重复序列,长度较短,且广泛均匀分布于真核生物基因组中(Chambersetal., 2000)。由于重复单位的次数不同或重复程度的不完全相同,造成了SSR长度的高度变异性,其中最常见的双核苷酸重复类型,如(CA) n(Jérmeetal., 2010)。本研究分析中采用MISA 1.0,默认参数;对应的各个unit size的最少重复次数分别为(1~10,2~6,3~5,4~5,5~5,6~5)对Unigene进行SSR检测,共检测到36 323个微卫星序列,发生率为61.30%(36 323/59 257)。其中单核苷酸重复、二核苷酸重复、三核苷酸重复、四核苷酸重复、五核苷酸重复、六核苷酸重复微卫星序列分别为21 508、9 217、5 290、261、22、25条,依次占微卫星序列的59.21%(21 508/36 323)、25.38%、14.56%、0.72%、0.06%、0.07%,其中单核苷酸重复占比最高,其次是二核苷酸和三核苷酸重复。并且对不同SSR类型在基因转录本的密度分布进行统计(图6)。

图4 麦红吸浆虫Unigenes的KEGG分类图Fig.4 KEGG functional categories of Sitodiplosis mosellana Unigenes

图5 麦红吸浆虫Unigenes的CDS的长度分布统计图Fig.5 Length distribution of CDS of Sitodiplosis mosellana unigenes

图6 麦红吸浆虫Unigene的不同SSR类型在基因转录本的密度分布Fig.6 Density distribution of Unigene SSR in different types gene transcripts of Sitodiplosis mosellana
3 结论与讨论
转录组学(Transcriptomics)是功能基因组学研究的一个重要内容,它是从整体水平上研究细胞中基因转录的情况及其转录调控规律。基于高通量测序技术的转录组测序(RNA-seq)通过对组织中的RNA(包括mRNA和非编码RNA)进行测序,能够全面快速地获得某一物种特殊组织或器官在某一特定状态下的几乎所有转录本信息,具有高准确性、高通量、高灵敏度和低运行成本等突出优势,已经广泛应用于各种生物转录组的研究(Shendure, 2008; Grabherretal., 2011; 张春兰等,2012)。为获得麦红吸浆虫的转录组信息,本研究应用Illumina高通量测序技术对麦红吸浆虫进行了转录组测序,通过序列的预处理Trinity拼接、 转录本功能注释等步骤,得到了数据量为27.88 G的转录组信息。大量的麦红吸浆虫转录本信息可为其今后的分子标记和功能基因挖掘提供有价值的信息。
通过高通量测序,经拼接组装,共获得77 500个转录本,进一步分析获得59 257个Unigenes,总长度49 861 164 bp,最短201 bp,最长29 282 bp,平均长度841 bp,N50值为1 802 bp(N50是指将拼接转录本按照长度从长到短排序,累加转录本的长度,到不小于总长50%的拼接转录本的长度),N50值越大,反映组装得到的长片段越多,组装效果就越好。测序数据产量和数据组装质量是转录组测序完成情况的重要指标。以上研究结果表明,此次序列组装的质量和长度可以满足转录组分析的基本要求,且新一代高通量测序技术是批量麦红吸浆虫功能基因的更为有效手段,进一步说明Illumina HiSeq 2000是高通量转录组测序的可靠平台。
结合生物信息学分析方法,对麦红吸浆虫Unigene与7个公共数据库(Nr,Nt,Pfam,KOG/KOG,Swiss-Prot,KEGG和GO)进行比对,以强大的生物信息学平台作支撑,根据“基因结构相似,功能同源”的原理,对基因的功能进行注释。分别获得18 596、2 868、9 110、14 680、18 912、19 584和11 279个结果,共计95 029个结果。表明在对麦红吸浆虫基因组不清楚的情况下,高通量测序技术是批量发现麦红吸浆虫功能基因的有效且简易手段。
本研究中利用Nr和SwissProt蛋白数据库对所获得的59 257个Unigenes进行BLASTX比对分析,共有30 088条序列可被编码,占全部基因的50.78%(30 088/59 257)。29 169个Unigenes与其它物种蛋白序列无匹配,占总体的49.22%,此部分Unigenes主要包括以下3种类型:(a)Unigenes序列片段长度过短,不能获得同源性比对结果;(b)基因注释信息的暂时缺乏。昆虫基因组学及转录组学研究刚刚起步,生物信息数据库仍在不断更新和完善中,基因功能注释信息不全会造成部分序列暂时无法获得对应的功能注释信息;(c)昆虫特有的新基因,可能存在一些昆虫特有的新基因,这或许也是导致其同源序列较难发现的原因之一。随着今后研究的深入,进一步将麦红吸浆虫与其它昆虫进行比较分析,为研究昆虫的分子生物学提供更为重要的信息。SSR分子标记具有操作简便、重复性好、多态性丰富、遗传信息量大和共显性遗传等优点,已在遗传多样性分析、遗传图谱构建、功能基因发掘、分子标记辅助育种等研究中得到了广泛应用(宋建等,2006;Yangetal., 2009; 刘峰等,2012)。采取实验室手段开发SSR引物费时、费力、成本高且试验复杂,基于转录组数据库信息进行SSR分子标记开发将是一种既经济又有效的方法。本研究通过查找发现了36 323个SSR位点,SSR不但出现频率高(发生率为61.30%),而且类型丰富。目前,已经有很多研究者通过转录组数据库发掘分子标记(Zhaietal., 2013; Lietal., 2016),解决了传统开发分子标记的方法耗时长、成本高的问题。本研究将为麦红吸浆虫后续分子鉴定、多态性检测和种群遗传结构的研究提供一定的参考价值。
本研究采用Illumina HiSeq 2000高通量测序技术建立了麦红吸浆虫转录组数据库,获得了大量的转录本信息,并对表达基因进行了序列组装、功能注释及代谢途径等分析,为今后更深入研究麦红吸浆虫功能基因组、基因克隆及麦红吸浆虫抗虫品种的研究提供了大量的信息资源,该转录组数据也还可以作为今后基因组的参考序列,为麦红吸浆虫的分子生物学研究提供宝贵的基因组数据来源。
