无催化剂合成芳基磺酰基吲哚的三组分反应*
2020-03-06蒲凡,陈博
蒲 凡,陈 博
(西华师范大学化学化工学院,四川 南充 637000)
吲哚化合物广泛存在于自然界中,而且临床医学发现由于它具有抗菌、抗肿瘤等生物活性,在生物医疗领域研究广泛,因此一直是国内外化学家研究开发利用的热点分子,吲哚环被称为芳香化合物的“指环王”。众所周知,砜具有抑制各种酶促过程和广谱医药应用的能力。因此,在吲哚体系中引入磺酰基官能团,形成3-(1-芳基磺酰基烷基)吲哚衍生物,是一种常见的做法[1-2]。此外,它们是有机合成中的多功能合成子,在广泛的合成转化方面有进一步的发展空间。芳基磺酰基吲哚通常通过吲哚与α-酰氨基砜的酸促进或催化Friedel-Crafts反应合成,α-酰氨基砜预先由氨基甲酸酯,醛和亚磺酸钠制备[3]。磺酰基杂环是预官能化的3-烷基吲哚与亚磺酸钠的亲核取代反应,其底物范围相当有限。不久前,Petrini等[4]发现了从简单的起始原料中通过多元组分反应(MCR)优雅直接合成3-(1-芳基磺酰基烷基)吲哚,但在这个缩合反应中,需要50mol%的布朗斯台德酸作为促进剂。鉴于磺酰吲哚化合物的重要性,仍然迫切需要制定温和经济的方案。一步和原子经济的方式,MCR是快速构建分子复杂性的有力工具[5]。由于富电子吲哚核表现出对亲电试剂的强的反应性,我们推测了亚磺酸尽管是一种弱酸,可能在吲哚MCRs中起到亲核试剂[6]和酸启动子的双重作用[7-8]。本文中,我们通过吲哚、羰基和苯亚磺酸的三组分反应提供了无催化剂的3-(1-芳基磺酰基烷基)吲哚的合成。
1 实 验
1.1 主要试剂与仪器
苯甲醛,吲哚,3-氯吲哚,4-甲氧基吲哚,4-氯吲哚,5-溴吲哚,5-硝基吲哚,苯亚磺酸钠,浓硫酸,甲苯,四氢呋喃,二氯乙烷,1,4二氧六环,二氯甲烷,乙酸乙酯,石油醚,碳酸氢钠,柱层析硅胶,薄层色谱硅胶板。除特殊说明外,所有溶剂均为国产分析纯溶剂,按常规方法干燥处理。
BioTOF Q高分辨质谱仪;Brucker-400 MHz型核磁共振仪;旋转蒸发仪;电热恒温水浴锅。
1.2 磺酰基吲哚化合物的制备方法
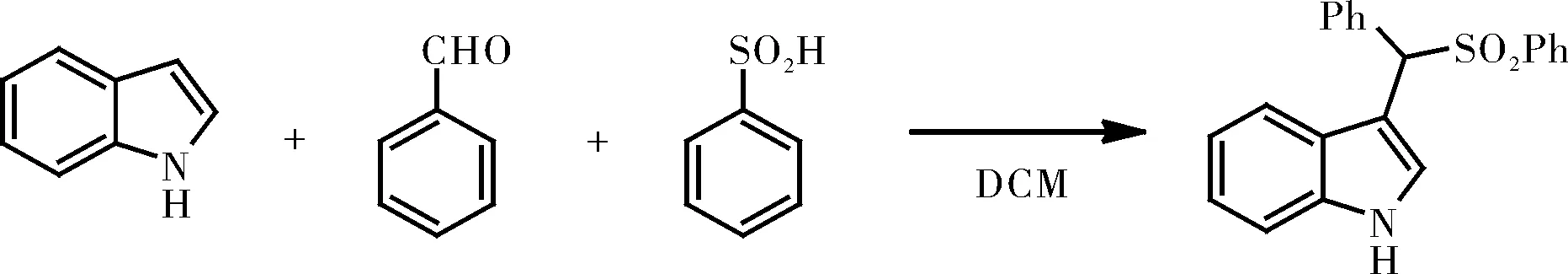
图1 芳基磺酰基吲哚的制备Fig.1 Synthesis of 3-(1-Arylsulfonylalkyl)indoles
取一个干燥干净的150 mL圆底烧瓶,加入磁力搅拌子和称取5.8 g无水苯亚磺酸钠,加入50 mL水搅拌完全溶解,冰水浴小心缓慢加入5 mL分析纯浓硫酸,搅拌15 min。冷却至室温停止反应析出大量白色固体,用二氯甲烷萃取直到水相澄清,加入无水硫酸钠干燥,静止过滤旋转蒸发仪旋干得到白色固体,真空干燥箱干燥后直接用于下一步。取干燥干净的50 mL 圆底烧瓶,烧瓶中装入磁力搅拌子和苯亚磺酸(624 mg,4.0 mmol),然后用注射器加入CH2Cl2(15 mL),将混合物搅拌10 min以完全溶解后,加入5-溴吲哚(392 mg,2.0 mmol),将苯甲醛(0.224 mL,2.2 mmol)用注射器慢慢滴加到反应体系,并将所得溶液在室温下剧烈搅拌2 h。TLC监测反应进行,原料5-溴吲哚完全消失后,停止反应,将反应混合物用5 mL饱和NaHCO3水溶液淬灭直到溶液至中性,然后用CH2Cl2萃取3次,饱和食盐水洗,无水硫酸钠干燥,旋转蒸发仪浓缩旋干并通过硅胶柱色谱分离纯化,洗脱剂石油醚和乙酸乙酯比例为5:1,浓缩旋干得到粉红色晶体678 mg, 产率86%。1H NMR(400 MHz, CDCl3): δ 5.62(s, 1H), 7.17(dd,J=1.6, 8.4 Hz, 1H), 7.25(d,J=8.0 Hz, 2H), 7.28~7.34(m, 4H), 7.56~7.61(m, 4H), 7.75(d,J=2.4 Hz, 1H), 7.83(d,J=2.01 Hz, 1H), 8.37(s, 2H);13C NMR(100 MHz, DMSO): δ 144.5, 135.9, 134.5,131.4, 126.7, 128.7, 128.5, 127.6,126.5, 125.6, 125.1, 116.1, 60.1. HRMS(ESI-TOF) Calcd for C21H16BrNO2S[M+H]+:426.3280. Found : 426.3286。
2 结果与讨论

表1 反应条件的筛选
注:a:The reaction was carried out using p-toluenesulfinic acid(0.2 mmol),indole (19.6 mg, 0.1 mmol) and benzaldehyde (0.11 mmol) in 1 mL DCM for 4~6 h。
由表1数据可以看出,我们使用过量的对甲苯亚磺酸,使用二氯甲烷做溶剂考察了温度对反应的影响,我们发现随着温度升高产率明显上升。当温度为10 ℃时,产率只有45%。原因可能是苯甲醛中的碳氧双键反应活性不好,需要升高温度来使苯甲醛活化,反应更容易进行完全,所以温度越低产率越低。当温度继续升高,苯甲醛慢慢氧化为苯甲酸,反应活性降低反应产率降低。最后,我们选择30 ℃为最佳反应温度考察其他条件的影响。

表2 溶剂对反应的影响
注:b:The reaction was carried out using p-toluenesulfinic acid (0.2 mmol),indole (196 mg, 0.1 mmol) and benzaldehyde (0.11 mmol) in 2 mL solvent for 4~6 h.
由于最佳反应温度为30 ℃,且近乎定量的产率。所以我们稍微筛选了一些比较常用的溶剂。含卤素溶剂:1,2-二氯乙烷能得到不错的收率;而醚类溶剂:1,4-二氧六环则只能得到中等偏上的收率,四氢呋喃产率比较低。但是DMF做溶剂得不到目标产物(entry 3),我们认为其原因是DMF中含羰基,会与吲哚发生反应,得到加成产物所以得不到目标产物。因此我们选择最优反应条件为:吲哚(1.0 eq),对甲苯亚磺酸(2.0 eq),苯甲醛(1.1 eq),二氯甲烷为溶剂,30 ℃反应。


表3 底物的拓展
The reaction was carried out using p-toluenesulfinic acid (2.0 eq),indole (1.0 eq) and benzaldehyde (1.1 eq) in 15 mL DCM for 4~6 h。
通过对不同取代基的考察,我们可以发现,不管是吸电子基还是供电子基,都能得到目标化合物其产率都很高;吲哚环上溴和甲氧基取代基的产率都是接近85%,可能是卤素溴对它的空间位阻的影响,而硝基则是吸电子基团,因为诱导效应反应受到影响。幸运的是,不管是吸电子还是供电子取代基均不影响反应的产率,均得到目标产物。
3 结 论
我们开发了一种无催化剂作用通过吲哚羰基和亚磺酸的MCR合成3-(1-芳基磺酰基烷基)吲哚的有效方法,该反应条件温和,操作简单成本低,并且产率非常不错,具有潜在有机合成价值和生物医学价值。本实验室目前正在对该化合物在天然产物不对称有机合成中的应用和两性离子π-烯丙基钯配合物的碳负离子从乙烯基环丙烷到原位形成的不饱和亚胺的共轭加成进行进一步的研究。
