盐酸帕洛诺司琼的合成工艺研究
2020-03-06李学强魏梦雪
全 军,李学强,魏梦雪
(1 上海华源药业(宁夏)沙赛制药有限公司,宁夏 银川 750002;2 宁夏天然药物工程技术研究中心,宁夏 银川 750021;3 省部共建煤炭高效利用与绿色化工国家重点实验室,宁夏大学化学化工学院,宁夏 银川 750021)
盐酸帕洛诺司琼,化学名为(3aS)-2-[(S)-1-氮杂双环[2.2.2]辛-3-基]-2,3,3a,4,5,6-六氢-1-氧-1H-苯[de]异喹林盐酸盐。盐酸帕洛诺司琼是MGI Pharma公司开发的5-HT3受体拮抗剂,临床用于治疗由化疗、放疗引起的恶心、呕吐等副作用,剂型为小水针,规格为5 mL:0.25 mg。首先在欧洲上市,商品名为Onicit,在意大利是由Italfarmaco公司负责销售。2002年9月厂商向FDA申报,2003年7月美国FDA批准上市,商品名为Aloxi Injection。有关盐酸帕洛诺司琼的合成工艺,经文献查阅主要有如下两条合成路线[1-2]:
路线一如图1所示,1,8-萘二甲酸酐与S-3-氨基奎宁环胺盐酸盐反应得酰亚胺中间体2,加氢还原后得到中间体3,化合物3在质子酸条件下发生消去反应得中间体4,化合物4加氢还原、提纯、成盐得目标化合物盐酸帕洛诺司琼1。在临床前研究过程中,我们采用本条路线进行样品的制备,该工艺制得的盐酸帕洛诺司琼的总收率为12.9%。
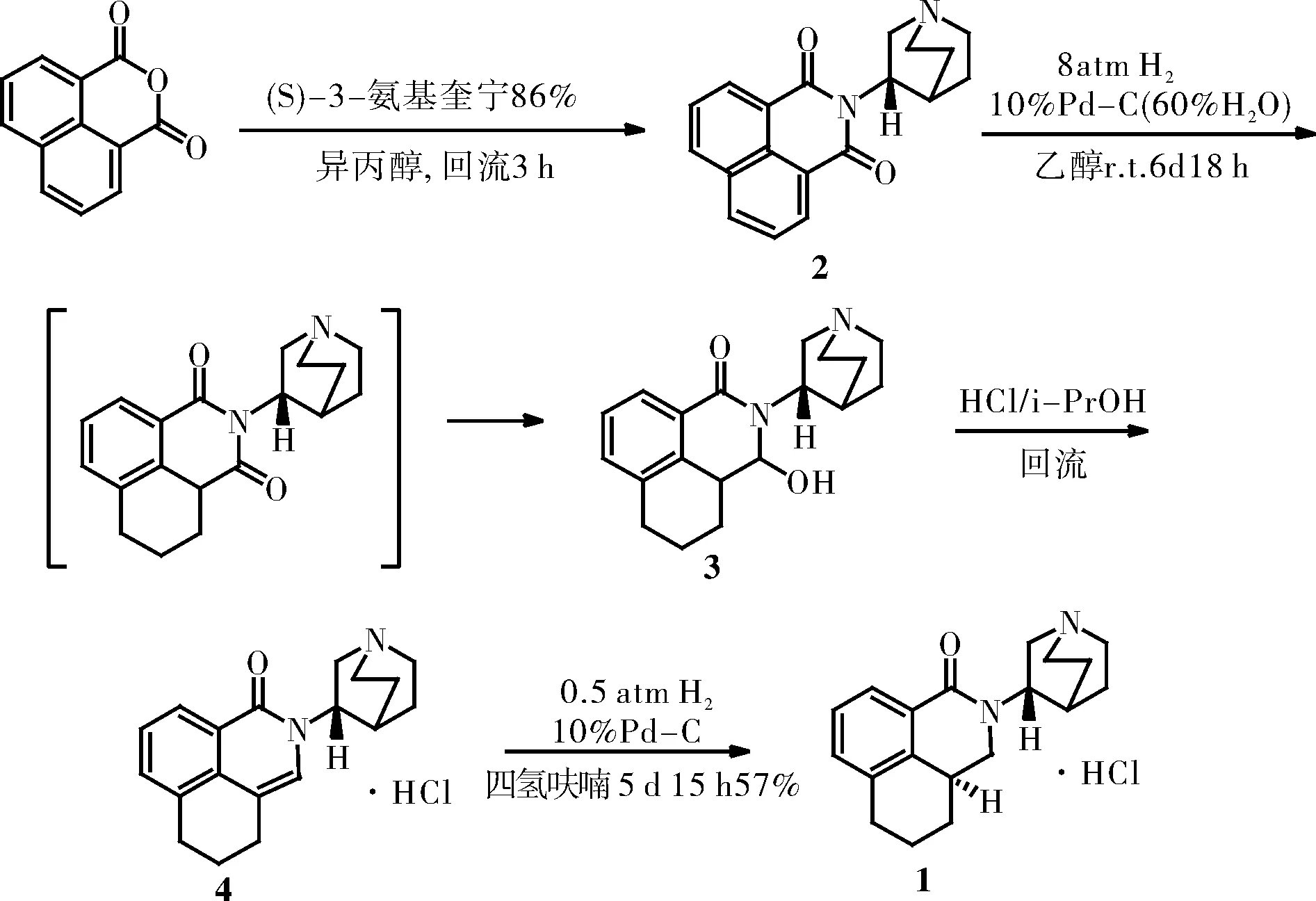
图1 盐酸帕洛诺司琼合成路线(1)Fig.1 Synthetic route of palonosetron hydrochloride (1)
路线二如图2所示。以S-四氢萘甲酸为起始原料与二氯亚砜反应生成酰氯,再与S-3-氨基奎宁环胺盐酸盐反应得酰胺中间体化合物(Ⅰ),中间体化合物(Ⅰ)经硼氢化钠还原成中间体化合物(Ⅱ),化合物(Ⅱ)经环合、成盐得目标化合物盐酸帕洛诺司琼。该工艺由S-四氢萘甲酸制备盐酸帕洛诺司琼的总收率为13.9%。

图2 盐酸帕洛诺司琼合成路线(2)Fig.2 Synthetic route of palonosetron hydrochloride (2)
经过文献检索,对以上两条路线进行比较与制备条件探索,发现两路线的起始原料均在市面上有售,比较两种合成路线,路线二较路线一操作更简单,反应时间更短,不用加压,安全性高,收率高,成本低。因此我公司参照文献对路线二进行了合成工艺研究,并结合自身条件对工艺进行优化,其中对中间体(I)的制备进行了条件改进和优化,体系中加入三乙胺缚酸剂后,酰胺化反应收率明显提高,产率由原来的50.4%提高到60.1%,最终确定了本品的合成路线,工艺的流程图见图3。
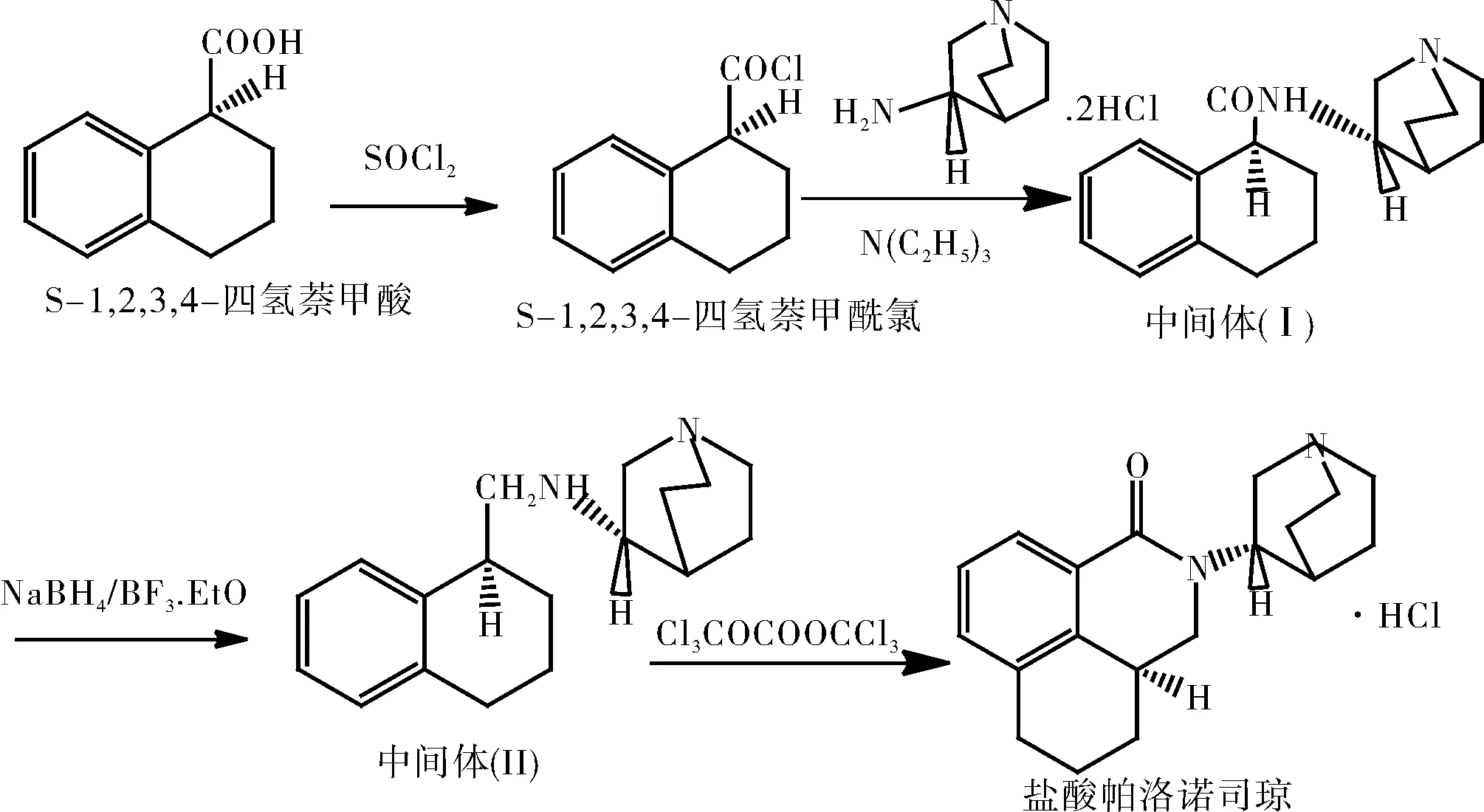
图3 优化后盐酸帕洛诺司琼合成路线Fig.3 Optimized synthesis route of palonosetron hydrochloride
1 实 验
1.1 试剂和仪器
Bruker Avance 400 MHz核磁共振仪(TMS为内标,CDCl3为溶剂);API QSTAR Pulsar飞行时间高分辨质谱仪(ESI源上测定);FTIR-8400S型红外光谱仪,日本岛津。
四氢萘甲酸,成都新恒创药业有限公司;氨基奎宁,成都新恒创药业有限公司;常规色谱柱硅胶(200~300目),青岛海洋化工厂;其余所用试剂均为分析纯。
1.2 合成工艺步骤
1.2.1 N-(1-氮杂双环[2.2.2]辛-3S-基)-1,2,3,4-四氢萘-1S-甲酰胺的合成
将42 g,0.239 mol (S)-1,2,3,4-四氢-1-萘甲酸加入到1000 mL的圆底烧瓶中,加入210 mL甲苯,搅拌得浅黄色澄清溶液,加入1 mL DMF,于室温下加入167 mL SOCl2,反应物在室温下搅拌反应2 h,然后在50 ℃搅拌反应2 h,50 ℃减压浓缩至干,加40 mL甲苯复溶,得到(S)-1,2,3,4-四氢-1-萘甲酰氯的甲苯溶液。
室温下,将47.8 g,0.239 mol (S)-(-)-3-氨基奎宁环胺盐酸盐悬浮于400 mL的乙酸乙酯中,同时体系中加入0.3 mol的三乙胺,然后将酰氯的甲苯溶液在室温下滴加到氨基奎宁环胺盐酸盐的乙酸乙酯溶液中,室温下搅拌反应1 h,再升温于50 ℃搅拌反应4 h。反应液用冰水浴冷却,向反应液中加入30% NaOH水溶液调pH至10,混合物用氯仿萃取100 mL×3,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏去除溶剂,得固体物,用甲苯冲结晶,干燥得到中间体(Ⅰ)酰胺34.2 g,mp:188.3~189.3 ℃,收率为60.1%。
1.2.2 N-(1-氮杂双环[2.2.2]辛-3S-基)(1,2,3,4-四氢萘-1S-甲基)胺的合成
将34.2 g,0.1203 mol中间体N-氮杂双环[2.2.2]辛-3S-基)-1,2,3,4-四氢萘-1S-甲酰胺悬浮在400 mL的四氢呋喃中,一次性加入18.8 g NaBH4,室温下缓慢滴加三氟化硼的乙醚溶液(8 M,111 mL,0.903 mol),控制反应液在室温下搅拌反应1 h,再升温回流反应3 h。冷却至室温,加入6 mol/L盐酸调pH值为1,回流反应2 h,减压浓缩至干,得胶状物。加入6 mol/L盐酸1700 mL,加热回流至澄清。冰浴冷却,用饱和NaOH水溶液调pH至12,混合物用氯仿萃取200 mL×3,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏去除溶剂,得中间体(Ⅱ)橙黄色油状物36.8 g。
1.2.3 盐酸帕洛诺司琼的合成
将还原胺中间体N-(1-氮杂双环[2.2.2]辛-3S-基)(1,2,3,4-四氢萘-1S-甲基)胺36.8 g,0.136 mol溶解于新蒸馏的甲苯中,滴加三光气36.2 g的甲苯溶液,氮气保护下于室温反应24 h,产生大量白色沉淀。加入三氟化硼的乙醚溶液(8 M,120 mL,0.975 mol),反应液变澄清,有油状物附于瓶底,反应物回流反应6 h,冷却至室温。
加入2 mol/L盐酸180 mL,混合物回流反应1 h,冰水浴冷却,分去甲苯层,水层用50% NaOH水溶液调pH到12,用氯仿萃取300 mL×3,合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏去除溶剂,得棕色油状物24 g。
将油状物溶于80 mL异丙醇中,加入饱和氯化氢的异丙醇溶液,析出固体,搅拌1 h,固体析出完全后,过滤,干燥,得类白色固体17.4 g。
固体加100 mL异丙醇重结晶后,过滤,干燥得13.5 g类白色结晶性固体,收率为33.7%,mp:302 ℃,1H NMR (400 MHz, Chloroform-d) δ 12.02 (s, 1H), 7.84 (q,J=4.0, 3.4 Hz, 1H), 7.36~7.21 (m, 2H), 4.95 (s, 1H), 4.02~3.69 (m, 4H), 3.34 (ddd,J=33.2, 25.3, 12.1 Hz, 4H), 3.18~3.00 (m, 1H), 2.85 (tdd,J=24.3, 16.1, 10.3 Hz, 3H), 2.34 (s, 1H), 2.30~1.86 (m, 6H), 1.76 (qd,J=13.0, 5.5 Hz, 1H).13C NMR (101 MHz, Chloroform-d) δ 166.23, 137.13, 135.15, 132.88, 128.40, 126.81, 126.20, 77.37, 49.43, 49.40, 49.35, 46.17, 35.03, 28.19, 26.13, 25.77, 24.36, 21.94, 19.83。
2 结 论
(1)对盐酸帕洛诺司琼合成工艺进行了以有机碱三乙胺作为缚酸剂,对关中间体酰胺产物化合物(I)的合成进行了优化和改进;
(2)中间体四氢奈甲酸酰胺奎宁化合物(I)的收率由原工艺路线的50.4%提高到60.1%,产品总收率由原来的13.9%提高到16.2%,生产成本明显降低。
