不同化学法分离解聚过程对木质素结构的影响
2020-03-05李小玉李广慈李学兵
李小玉,李广慈,李学兵
(1.中国科学院青岛生物能源与过程研究所生物燃料重点实验室,山东青岛266101;2.中国科学院大学,北京100049)
高速发展的现代工业急剧消耗化石燃料资源,同时引发气候变化、空气污染、酸雨等一系列环境问题,阻碍未来社会实现高效、清洁、经济、安全的目标[1-2]。为了减少对化石燃料的依赖,当前最需要做的是寻找新型能源替代不可再生的化石能源,该替代品至少应该具备以下特征:可持续发展、碳中性、广泛可得、不与食品生产相竞争[3]。和化石燃料资源相比,生物质种类多且分布广,主要包括动植物和微生物的残体及其代谢物等。其中植物中的木质纤维素是极少数能满足以上特征的资源之一,目前利用木质纤维素制备燃料和化学品已成为研究热点[4]。
木质纤维素由纤维素、半纤维素、木质素组成,其中木质素呈现复杂的三维网状结构,是纤维素和半纤维素的支撑骨架,是潜在的可持续能源以及大众化学品的重要原料来源[5]。木质素几乎占陆地生物量的25%,因此被认为是继纤维素之后第二大可再生资源[6]。目前,造纸和纸浆工业每年产生超过5 000万t的木质素,但绝大部分随着造浆废液被排放。工业纤维素生物乙醇行业每年还产生数百万吨木质素作为低附加值副产品。到2022年底,仅美国生物乙醇工业预计每年可产生高达6 000万t的木质素[7-8]。如果木质素的价值能够得到充分发挥,将木质素作为工业原料进行研究和应用,有望成为生产芳烃、环烷烃、烷烃等液体燃料和酚类精细化学品的重要可再生原料,其经济可行性和环境可持续性将会显著提高。
与纤维素和半纤维素相比,木质素的碳氧物质的量比(n(C)/n(O))最高,虽然其质量分数仅在15%~30%,能量储存却是高达40%以上[9]。木质素具备高热含量,但同时因其结构复杂性,它也是植物细胞壁中非常难降解的成分。许多研究证实,木质素通常用以下三类常规技术进行开发利用:(1)木质素的分离;(2)木质素的解聚;(3)木质素解聚产品的整合分析[10]。贯穿这三类技术工艺的一个很重要问题是木质素的结构不稳定性,所以在绪论部分,将主要从木质素结构的角度展开论述,并针对木质素加工过程中产生的各种结构变化提出促进木质素解聚反应的结构特征以及改善并稳定木质素结构的有效途径。
1 木质素资源概述
1.1 木质素来源
木质纤维素是一种主要由纤维素(质量分数为40%~60%)、半纤维素(质量分数为10%~40%)和木质素(质量分数为15%~30%)构成的复合高聚物(如图1(a)所示),主要存在于木质(杨树、桦树)和草本生物质(柳枝稷、芒草、玉米秸秆)中[10-11],由于木质素复杂的结构成分被称为超分子自组装聚合体[7,12-13]。木质素是通过植物细胞壁的单木质醇的自由基聚合形成的不规则三维高分子聚合物,它在植物细胞壁中相当于刚性黏合剂的作用,有助于细胞壁和纤维内部输送水分和养分,保护它们免受微生物侵袭,并提供机械支持,但同时也增加了其复杂程度,使木质素的降解工艺开发没有纤维素和半纤维素那么成熟[10,14]。
1.2 木质素天然结构组成
木质素生物合成来源于苯丙烷单元,是一种多酶催化的生物化学网络,其中苯丙氨酸和酪氨酸被转化为主要的木质素结构单元:对-香豆醇、松柏醇和芥子醇。这些结构单元或单木质醇主要区别是酚核上的甲氧基量不同(如图1(b)所示),通常缩写为H(对羟基苯基)、G(愈创木基)和S(丁香基)[15]。木质素中酚核的相对分布因植物物种的不同而存在很大区别。通常,软木木质素(例如松树、云杉)仅含有G结构单位,而硬木木质素(例如桦树、杨树、桉树)由G和S结构单元组成。来自草本生物质的木质素含有H、G、S 3种结构单元,但是H结构单元质量分数通常较低(<5%)[16-17]。除了这3种主要结构单元,木质素还存在其他(酚类)化合物(如图2所示),例如在木质素的侧链α位或γ位上,存在对羟基安息香酸、香草酸、紫丁香酸、对羟基肉桂酸、阿魏酸等酯类,除了酯型结构,还存在醚型联接,同时存在碳键联接。侧链结构的差异性对其他的化学反应性能产生很大影响[14,18]。
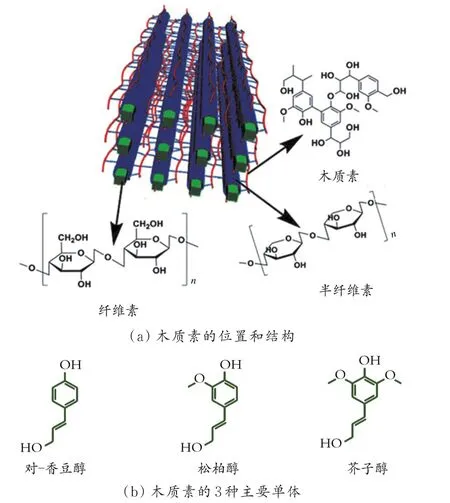
图1 木质纤维素生物质中木质素的位置、结构和3种主要单体[6]

图2 木质素主要结构单元以及化学键组成[14]
在木质素生物合成过程中,发生木质化反应(自由基聚合),产生各种醚和C-C单元间键。木质化由漆酶和过氧化物酶引发,通过酚羟基和苯氧基两类基团进行聚合形成木质素大分子,进而通过共价键和非共价键与纤维素和半纤维素发生化学相互作用,形成复杂的三维异质网络[19-20]。由于单木质素的共轭π-体系,可以形成各种单元间联接,包括醚键和C-C。最丰富且众所周知的联接类型是β-O-4,其数量在几类典型的化学键中(β-O-4,β-5,β-1,5-5,α-O-4,4-O-5,β-β)占50%以上,与α-O-4(在苯基香豆素和二苯并二氧辛环素结构中)联接一起[21],这些醚键最容易裂解,因此它们通常是木质素解聚方法的主要目标。与碳水化合物聚合物相比,木质素还含有相当大比例的C-C单元间键,如5-5(在二苯并二氧辛环和联苯中)、β-5(在苯基香豆素中)、β-1(在螺噻二烯酮中)和β-β(树脂醇)联接[22-23]。天然木质素中C-C单元间键的相对数量受单木质醇分布(即n(S)/n(G)/n(H))的强烈支配,甲氧基取代的邻位不能参与5-5和β-5的C-C的形成[24-26]。因此,主要由S-单元(硬木)组成的木质素含有比由G-单元(软木)组成的木质素更低的C-C含量,这些差异对木质素解聚作用产生重大影响。
2 木质素解聚效率低的原因分析
2.1 木质素分离过程中结构变化
木质素的多种化学键的解离存在很大差别,如图3所示。总的来说,C-C的解离能要高于C-O,在C-C中,β-5的解离能最高[27],因此C-O醚键的数量越多,木质素解聚效果就越好,而其中的β-O-4在总化学键中最为丰富,所以单体产量很大程度上取决于木质素中β-O-4的数量。其中,β-O-4可以通过包括水解在内的多种方法进行解离,例如利用酸碱催化、金属催化剂或者氧化的方法,但是相对顽固的C-C的断键较为困难[28]。受坚固的C-C的限制,导致不同质地木材中的木质素的理论单体收率存在明显差异,其中硬木中木质素收率约45%,而软木中仅为20%左右,且很容易在大多数预处理或者解聚阶段分解[29]。因此,分离木质素很大程度上取决于所用分离方法的类型和工艺条件的苛刻程度。

图3 天然木质素和工业木质素中联接(β-O-4和C-C)分布的示意图[30]
在分离过程中很难保持木质素的天然结构。木质素经过一系列的解聚和再聚合反应,使处理后的木质素与天然木质素相比,β-O-4和C-C含量已经发生了巨大的变化[30-31](如图4所示)。在木质素分离过程中,使用催化剂(酸或碱)会引起木质素缩合反应,形成额外的、更难分解的C-C,对工业木质素的HSQC核磁共振研究表明,木质素单体的特征侧链,如α-羟基和β-O-4,由于复杂的缩合反应迅速减少[32]。保留β-O-4的木质素也不能保证在后续氢解反应中得到单体,因为β-O-4的部分可能与其他部分通过C-C相连[33],使它们受到周围基团产生的空间位阻效应的保护,进而很难在多相催化剂表面吸附[34](如图4所示)。此外,木质素与催化剂之间的传质受到溶剂的影响,一般情况下,木质素在溶剂中呈现悬浊液,解聚过程中不利于扩散和传质[35],理想情况下,就解聚效率而言,均相木质素流是最好的,但是目前很难达到该理想状态。
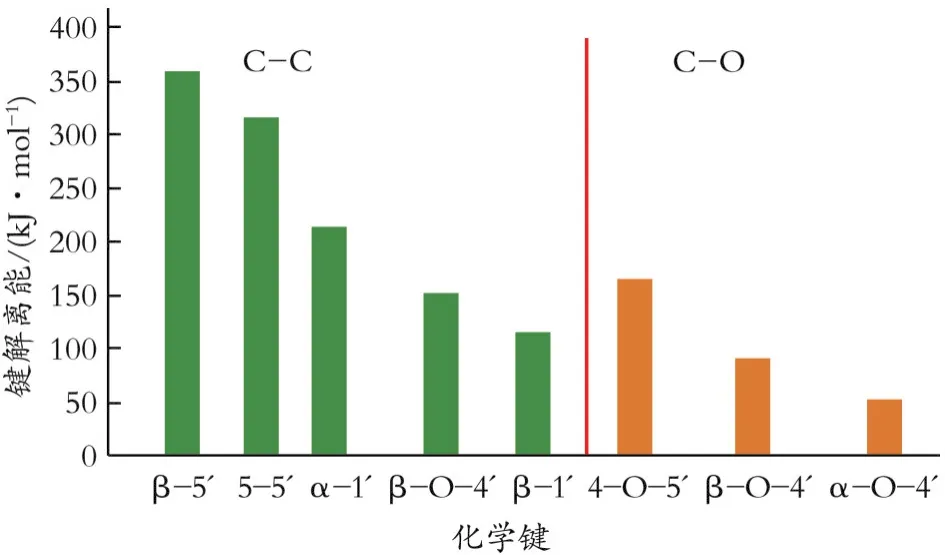
图4 木质素C-O和C-C的平均键解离能(BDE)(对模型化合物计算)[34]
为了将木质素从纤维素和半纤维素中高效剥离,近年来研究者们已经开发了多种加工方法来分离木质素。由于木质素特殊的热力学性质,每种方法都需要在试剂和所需能量方面进行权衡,这不仅影响最终产品的分子量和纯度,还会在分离阶段引入新的化学官能团。因此,深入认识木质素的化学性质在分离提取过程中产生的变化,对于木质素后续的开发利用非常重要。
2.1.1 稀酸分离法 木质素可以通过磷酸二铵(DAP)、重水磷酸氢钾(HWP)、过氧化苯甲酰(SEP)等进行处理[28]。这一过程在高温(165~195oC)下进行,持续时间为3~12 min,随后通过水洗工序[36]。该方法可在相对温和的酸性条件下获得具有高溶解度的木质素[37-38],但是反应的总转化率较低,为0.5%~1.4%,并且得到的木质素含有糖衍生的杂质,具体原因与酸处理过程中结构的巨大改变有关。酸浓度和温度越高,木质素结构的变化程度越大,越易发生生物量迁移和再分布现象。
在酸处理条件下,木质素主要发生芳醚键酸解和酸催化缩合两类反应[39]。木质素中的β-O-4型醚键遇酸易水解,而木质素大分子内的碳正离子中间体与亲核试剂反应会导致木质素发生再聚合反应(如图5所示)。经稀酸预处理后的木质素中β-O-4含量通常较低,R.Samuel等[40]报道,稀酸预处理使柳枝稷木质素中β-O-4减少近36%。另有报道称,在稀酸预处理条件下,紫丁香基单元比愈创木基单元更容易发生降解反应,导致预处理生物质中木质素的n(S)/n(G)降低。此外,稀酸预处理导致新的酚羟基形成,部分原因是β-O-4型醚键断裂,同时也观察到其他结构变化,如甲氧基含量降低、乙酰基水解出现新的肉桂醛单元和众多很难处理的C-C[41]。
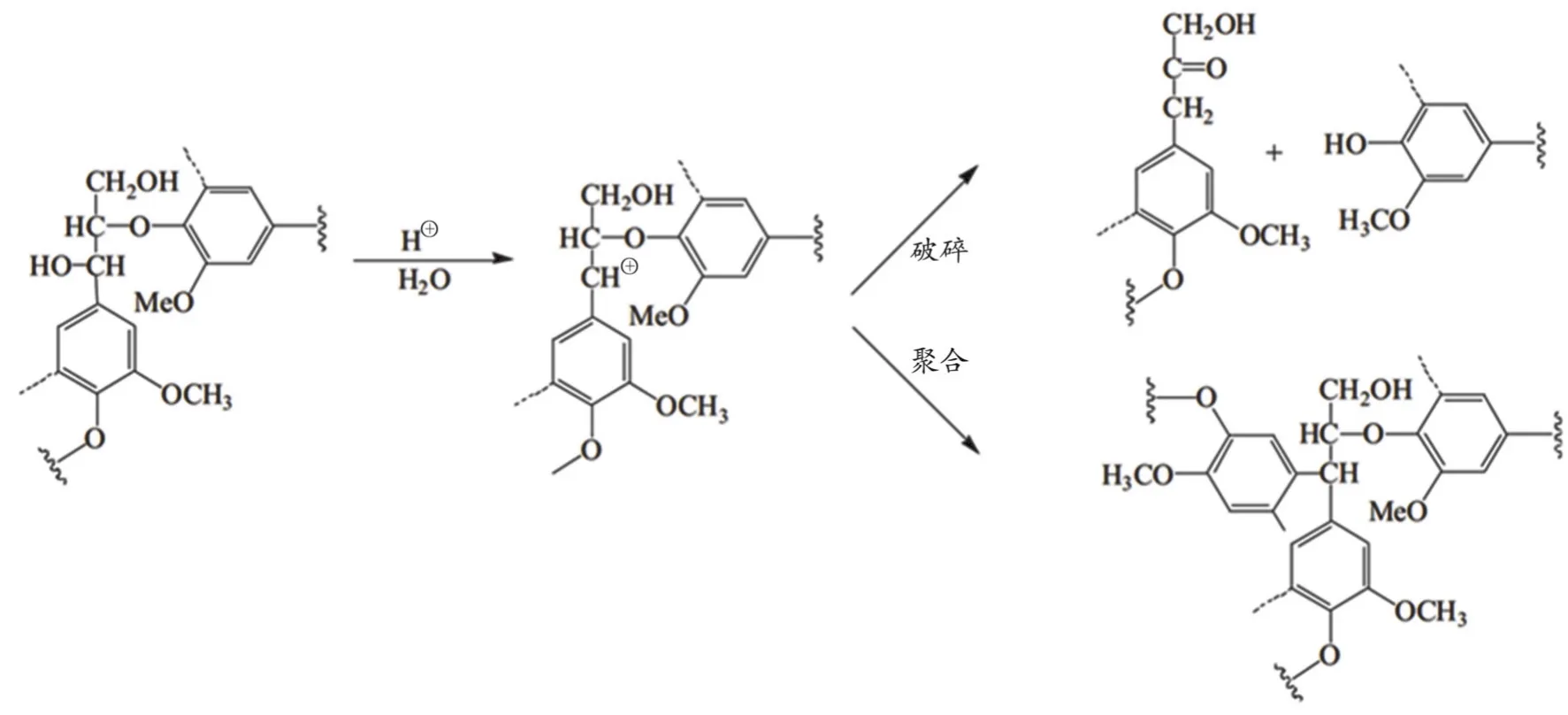
图5 酸处理过程中β-O-4单元的结构变化[36]
2.1.2 碱性分离法 在硫酸盐制浆过程中,木材在NaOH和Na2S的水溶液中加工,其HS-通过脱木质素和木质素解聚来提高制浆的选择性,而不会同时加速碳水化合物的溶解[42]。然而,苛刻的碱性环境会导致木质素严重降解和再聚合反应。此种木质素含有少量残留的β-O-4,还以硫醇基的形式引入硫,这使下游的还原过程复杂化(例如催化剂中毒)[43]。
亚硫酸盐制浆的基本过程是木材或生物质与亚硫酸钙/镁等在125~150℃的高温下反应3~7 h[35,44-45]。根据选择的亚硫酸盐种类,体系的酸碱度可以从2到12不等。选择合适的亚硫酸盐类在碱性环境下分离木质素,β-O-4和α-O-4都会发生断裂,并在木质素主链上添加磺酸基,引入极性基团到木质素骨架有助于其在蒸煮液中的溶解和降解[46-47]。木质素溶解,其中纤维素部分保持固体,生成的木质素磺酸盐的相对分子质量较大,数值为1 000~50 000[48]。碱处理过程也使木质素发生很大变化[49-50]:(1)发生了许多缩合反应,导致形成新的C-C(方案1);(2)与硫酸盐制浆法引入的硫质量分数相比,磺酸盐形式存在的硫质量分数更高(3%~8%);(3)木质素磺酸盐含有大量非木质素物质,其中高达30%的碳水化合物、灰或其他杂质,这些改变使木质素的分离和下游产品更加复杂化。
图4揭示了两个重要的观察结果[45-51]:(1)木质素缩合物具有比苄基阳离子更低的活化能,表明木质素缩合在苄基阳离子形成后自发地进行;(2)木质素缩合具有比木质素水解更低的活化能,表明木质素缩合比其水解进行得更快。这两个发现表明,在酸分馏过程中大量冷凝是不可避免的。理论上,降低温度和/或降低酸负荷将减少冷凝,同时还会破坏β-O-4,这会危害木质素的去除和溶解性。
2.1.3 有机溶剂法 有机溶剂法是以甲醇、乙醇、乙酸、过氧甲酸等有机溶剂或它们的水溶液在120~200℃的条件下对生物质进行处理的一种方法[44]。与其他方法相比,有机溶剂法的反应介质可以有效地溶解木质素,而不会对其结构进行较大程度的改变[52]。尽管已经证明了提取过程中可以进行自催化,但是该方法可以与催化剂(通常是碱或酸)结合使用[53]。在后一种情况下,可以分离出相对清洁且不含硫的木质素[54]。在制浆期间,主要是木质纤维聚合物中的碳水化合物和α-O-4被破坏。与其他制浆方法相比,β-O-4的裂解发生的程度较小,有机溶剂制浆可以被认为是在提取过程中保持木质素天然结构的更有效的选择[55]。通常,有机溶剂木质素具有更低的相对分子质量、更高的不饱和度,可以更好地保留木质素的天然结构,比脱碱木素和木质素磺酸钠更容易发生解聚[56-57]。但是,在某些特定条件下会发生一些变化,例如在酸性条件下,有机溶剂木质素的随机缩合反应与解聚反应会出现相互竞争。这是由于酸性条件能够促进苄基阳离子的形成,这些阳离子又与富电子芳环发生反应形成新的C-C,进而促进木质素的缩合[58]。此外,提取的木质素结构还会因为醇分子的亲和作用而产生部分侧链出现烷氧基化的现象,进而影响后续的解聚效率[59]。
上述三类化学制浆法原理是溶解纤维壁中的大部分木质素,纤维素和半纤维素通常作为固体保留在纤维壁上。在制浆过程中,木质素发生结构重排,与脱木素试剂(如硫化物和亚硫酸盐)发生亲核反应或木质素分子之间发生反应。此外,在这一过程中还发生了其他类型的C-O和C-C裂解反应。木质素片段被转化为一个自由基或离子物种,进而发生随机重组,形成非自然的C-C,最终形成全新的三维结构。
2.2 木质素解聚过程中的再聚合行为
木质素不仅在分离过程中经历着变化,在解聚反应中,反应条件(温度、压力、溶剂选择等)往往非常苛刻才能得到比较满意的效果[60]。原始木质素是非晶热塑性高聚物,存在大量酚羟基、醇羟基以及其他活性含氧官能团。在120℃下处理时,木质素通过醌甲基中间体脱除β-O-4中的γ-OH,生成甲醛。原始木质素也极易发生自聚反应,在高于其玻璃化转化温度(Tg)20℃以上加热20 min,平均相对分子质量增长近2个数量级[61]。不同预处理方式下得到的木质素其玻璃化转化温度也是存在很大差异,例如Kraft木质素Tg为124~174℃、苏打木质素Tg为 150~155℃、有机溶剂木质素(OSL)Tg为91~97℃。蒸汽爆炸所得木质素Tg为113~139℃。在木质素的各种解聚工艺中,反应温度远远高于其玻璃化转化温度,可以预见在反应条件下的木质素原料的自聚现象也将非常严重,特别是在木质素不良溶剂中进行的反应,木质素自身的这些物理化学性质导致其解聚效率偏低。
总之,木质素再聚合和木质素解聚是两个平行反应,两者都受反应温度和pH的影响。简单地优化反应参数并不能减少或消除木质素缩合,因此激发了研究人员对于新策略的探究。生物方法通常采用基因修饰的方式,例如对高紫丁香基生物质遗传修饰的成功表明,在木质纤维素生物合成期间阻断参与形成单元间C-C的反应性位点,是减少或阻止缩合反应的可行策略。化学方法主要是催化剂的开发以及结构改性。
3 木质素活性基团的钝化研究
目前,用于木质素解聚的方法主要分为生物法和化学法,生物法具有环保、选择性高的优点,但其解聚速度较慢、催化剂成本高且稳定性差,很难大规模工业应用。相对而言,化学法成本偏低、产物的选择性较高且易分离。所以,化学方法开发木质素是当今木质素研究领域的主流方向。现阶段,常见的化学法主要包括热裂解、水解、催化氧化、加氢解聚几大类,其中加氢解聚法是木质素转化常用方法之一。
相比之下,软木木质素显示出相对较高的玻璃化转化温度(Tg为138~160℃),最可能是由于更高量的分子间氢键和更大量的稠合结构,而硬木木质素显示出相对较低的玻璃化转化温度(Tg为110~130℃)。在分子模型上进行DFT计算,预测了β-O-4的键离解能(BDE)值为226~300 kJ/mol。值得注意的是,取代基效应可对β-O-4的BDE产生显著影响。例如,α-羟基氧化成酮可使BDE降低63 kJ/mol。由于木质素结构极其复杂,反应条件非常苛刻,非常容易出现过度加氢的问题,并且解聚过程中木质素本身之间以及活性反应中间体之间非常容易发生再聚合现象,生成焦油等更加难转化利用的物质,虽然发现了再聚合这一问题,但是对聚合机理还未得到满意的解释,使近几十年木质素高效利用技术的发展比较缓慢。针对上述问题,国内外许多研究人员做了以下工作。
X.Huang等[62]提出了乙醇在木质素解聚过程中,通过O-或C-烷基化反应对解聚产物进行封端以及作为活性产物甲醛的牺牲剂阻断解聚产物的再聚合反应,提高了酚类单体收率(60%~86%)。P.J.Deuss等[63]采用与二醇进行羟醛缩合、Ru/C催化原位加氢还原以及原位催化脱羰基的方式,有效弱化对羟基苯乙醛的反应活性,提高了木质素的解聚效率。采用原位加氢方法后的反应效果最为明显,液体收率达到95%以上,其中近40%为低相对分子质量解聚产物。J.Long等[64]以甲醇为溶剂进行木质素碱催化降解过程中,通过添加Ru/C催化剂原位氢解甲醇生成氢气,对酚类反应活性中间体催化加氢,在260℃实现了木质素的转化率达到90%以上,焦炭收率低于15%。R.Shu等[57]在研究Lewis酸以及Pd/C催化木质素加氢解聚过程中发现,木质素及其解聚产物在反应过程中的甲基化(methylation)能够有效地抑制解聚产物的再聚合反应。S.Dabral等[65]以CsCO3为催化剂,在碳酸二甲酯溶剂中进行木质素碱催化解聚。反应产物2DNMR(HSQC)的表征结果表明,在180℃条件下,溶剂型木质素的β-O-4以及4-O-5二聚单元中醚键完全消失;碳酸二甲酯在反应过程中既是溶剂也是反应试剂,将木质素基解聚产物的羟基进行甲基化钝化,避免了活性组分的再聚合反应。值得一提的是,以β-O-4二聚体为原料进行解聚反应时,解聚产物的化学计量不平衡,3,4-二甲氧基苯甲酯含量远远低于邻二甲氧基苯,这预示着部分产物进行了二次反应生成了聚合物。因此,尽管羟基的甲基化对木质素解聚产物起到一定钝化作用,但并未影响含醛基化合物的再聚合反应活性。J.A.Lercher课题组在相对温和条件下,以固体酸负载Ni为催化剂,在水相中对β-O-4以及α-O-4二聚体进行氢解,得到烃类化合物或者脂肪醇,其中醚键的氢解生成苯酚(环己醇)/烷基苯在120℃下即可反应完全[66-67]。J.Kong等[68]发现将上述催化体系应用于实际木质素的氢解,反应中仍会生成大量的焦炭/焦油。H.Konnerth等[69]以Ni7Au3纳米粒子为氢解催化剂,在水相中进行了木质素以及模型化合物的氢解,温和条件下(160℃、H2压力 1.0 MPa)发现NaOH的添加能明显地促进了木质素C-O氢解,并抑制了金属催化剂的苯环加氢能力,进而提高酚类产物的选择性,避免了氢解工艺中消耗大量氢。K.Parta等[70]以层状结构的水滑石焙烧后得到碱性多孔氧化物(Porous Metal Oxide,PMO)为载体,制备了负载型Cu/PMO催化剂,在温和条件下实现了木质素高效解聚,同时没有任何焦炭生成。C.Cui等[61]以硫酸二甲基为甲基化试剂,对原始木质素的酚羟基进行甲基化,改善其热稳定性。结果表明,完全甲基化的木质素在高于其玻璃化转化温度20℃下加热60 min,相对分子质量没有任何增加。S.Sen等[71]同样报道了使用碳酸二甲酯进行Kraft木质素甲基化的绿色方法。结果表明,纯木质素中存在的大量酚羟基通过形成分子内氢键而使其具有更高的Tg,从而产生物理交联结构。然而,随着木质素甲基化的降低,Kraft木质素的Tg也降低。完全甲基化后,未取代的木质素Tg从153℃降低至128℃,因为在完全衍生化后分子内氢键被消除。此外,甲基化可以消除自由基引发自聚合的可能性,使其成为天然聚合物商业加工的重要步骤。
4 结论与展望
国内外研究人员都旨在寻找一种温和、高效、绿色的木质素催化解聚工艺,提高催化解聚效率,明确催化解聚机理,但是目前解聚效率仍需提高,解聚和再聚合机理尚不明确。制约木质素高效利用的一个比较关键性的问题是木质素结构具有极大的不确定性,并且在木质素反应过程中,再聚合反应速率远远大于解聚反应速率。无论是在木质素分离过程,还是解聚过程,不同的处理工艺所得到的工业木质素与天然木质素都存在着很大的差异性。为了实现木质素的高效解聚,研究者在木质素分离和解聚过程中分别做了大量工作,但是综合性的研究很少,且选取木质素的来源不同,分离方法不同,解聚产物的评估定量方法也存在差异,相关数据比较的准确性有待提高。因此,为了指导和改进对木质素催化转化的研究工作,建议选取植物来源相同的木质素,采用统一的定量标准,将木质素整体作为研究对象,综合考虑木质素分离和木质素解聚,在分离过程中最大程度地保留木质素天然的β-O-4,减少C-C的生成,在解聚过程中减少活性含氧基团,稳定木质素解聚产物,减少再聚合反应的产生。将木质素结构最佳化,从而实现木质素的高效解聚。
