化学链燃烧中H2S与CuFe2O4载氧体的反应机理
2020-03-04方瑞雪
李 钰, 刘 晶, 刘 丰, 方瑞雪
(华中科技大学 煤燃烧国家重点实验室,湖北 武汉 430074)
化学链燃烧(Chemical-looping combustion, CLC)作为新一代洁净、高效的燃烧技术,可以实现CO2内分离和化学能的梯级利用,并避免NOx污染物的产生[1-2],已成为当前的研究热点。化学链燃烧技术包含2个串联的反应器:燃料反应器和空气反应器,通过固体载氧体在2个反应器之间往复循环,来实现氧的传递[3-5]。
载氧体作为化学链燃烧的关键[6],除了需要具备价格低廉、来源广泛和环境友好的特点外,还应具有良好的反应活性[7]和较高的燃料转化率[8-9]。目前常用的载氧体包括铜基和铁基等[10-11],其中铜基载氧体有较高的反应活性和载氧能力,无积炭发生,但由于Cu及其氧化物熔点较低,因此在高温下易烧结和团聚。相比之下,铁基载氧体具有环境友好、热力学稳定性好和机械性能优良等优点,但其反应活性和实际载氧量较低[12]。为了克服单金属载氧体的缺陷,双活性组分载氧体受到了越来越多的关注。实验表明,复合金属载氧体可以通过不同活性组分之间的协同作用和优势互补,具有很好的综合性能[13-14]。其中,具有尖晶石型结构的CuFe2O4广泛应用于污染物去除[15]、蒸汽重整[16]、燃料电池[17]和化学链燃烧[18]等领域。
CuFe2O4集合了铜基和铁基载氧体的优点并能克服单金属载氧体的缺点[19],既有良好的反应活性,又不容易发生烧结。Siriwardane等[20]的实验结果表明,少量的Fe2O3在温度约500 ℃就会与CuO形成尖晶石相的CuFe2O4,与相应单金属载氧体在化学链燃烧过程中的表现相比,CuFe2O4表现出更优的反应活性和结构稳定性。Wang等[21-22]考察了CuFe2O4载氧体与不同燃料在化学链燃烧过程中的反应性能,发现CuFe2O4载氧体相比单一金属载氧体(CuO和Fe2O3)具有巨大的优越性,它不仅具备良好的抗烧结性,而且燃料适应性较好。
H2S作为一种常见的有毒、腐蚀性可燃气体,不可避免地存在于煤制合成气和天然气原料气中。天然气通常含有少量H2S,约为20 μg/g。工业燃气中的H2S质量分数可根据现场情况而变化,在一些现场可达到800 μg/g,而对于未经处理的合成气,甚至可能大幅上升至8000 μg/g[23]。高正平等[24]采用实验方法研究了NiO载氧体与高浓度CO和H2S混合气的还原、硫化反应,结果表明:载氧体在还原反应过程中与H2S反应生成Ni3S2,导致载氧体表面发生烧结,进而显著降低了氧化还原过程的反应速率。Tian等[25]采用热重分析和X射线光电子能谱(XPS)的实验方法对H2S与铜基载氧体在化学链燃烧中的反应特征进行研究,结果表明:CuO载氧体的氧化还原能力受到H2S的影响而出现明显的下降,产物中检测到CuS。迄今为止,采用实验方法对H2S与载氧体在化学链燃烧过程中反应特性的研究较多[26-27],而对化学链燃烧过程中H2S与载氧体之间具体的相互作用机理尚不明确。因此,研究化学链燃烧过程中H2S与CuFe2O4载氧体的相互作用机理非常必要,可以为复合载氧体的应用提供理论指导。
笔者以CuFe2O4作为载氧体,采用密度泛函理论和周期性结构模型研究H2S在CuFe2O4载氧体表面的吸附机理。在探明CuFe2O4表面吸附位活性的基础上,进一步研究H2S与CuFe2O4载氧体之间的反应机理。
1 模型建立与计算方法
应用密度泛函理论对相关机理进行研究。计算过程中采用广义梯度近似方法(Generalized gradient approximation, GGA)描述电子交换关联势能,其中交换关联泛函为Perdew-Burke-Ernzerhof (PBE)泛函[28]。静电势考虑作用在系统价电子的有效势,选择超软赝势。电子波函数基于平面波基组展开,截断能设置为340 eV。布里渊区积分使用Monkhorst-Pack网格,表面吸附计算使用的k点为 4×4×1。结构优化计算参数设置:(1) 自洽场精度收敛标准为1×10-6eV/atom;(2) 能量收敛标准为1×10-5eV/atom;(3) 原子最大受力收敛标准为0.3 eV/nm;(4) 最大压力收敛标准为0.1 GPa;(5) 最大位移收敛标准为1×10-5nm。
自然界中CuFe2O4晶体一般以反尖晶石结构[29]存在。CuFe2O4晶体最稳定的低指数晶面为(100)面[30],其中稳定性较高的八面体端面由Cu、Fe、O原子构成,因此表面模型基于CuFe2O4(100)面的八面体端面构建。计算过程中的晶体模型及表面模型如图1所示。

图1 CuFe2O4晶体模型和表面模型Fig.1 The crystal models of CuFe2O4 and slab models of CuFe2O4 surface(a) Crystal model; (b) Slab model
表面结构采用具有5层原子层的周期性平板模型,其中固定最底层原子,上面4层原子完全弛豫以模拟实际表面,真空层厚度设为1.5 nm,用以防止平板间吸附质受到上层平板底部的影响。鉴于CuFe2O4是亚铁磁性材料,计算过程中四面体位金属原子与八面体位金属原子的电子自旋方向相反并设为高自旋态[31]。H2S分子放置在1 nm×1 nm×1 nm的正方体内进行结构优化。
优化后,晶体模型的晶格参数(a=c=82.21 nm,b=88.73 nm)与实验值(a=c=82.16 nm,b=87.09 nm)[10]较吻合,说明了计算方法的可靠性,因此建立的模型可以用来模拟CuFe2O4表面。
吸附能是表征吸附质与表面相互作用强弱的重要参数。吸附能(Eads,kJ/mol)的定义如式(1)所示,它表示吸附前后各物质总能量的变化。
Eads=E(sor-sur)-Esor-Esur
(1)
式(1)中,E(sor-sur)、Esor和Esur分别为吸附质在表面吸附后体系的总能量、吸附质的能量和表面的能量,kJ/mol。Eads为负数表示吸附过程为放热过程,且负数绝对值越大说明吸附作用越强。
过渡态指的是势能面上基元反应路径中的能量最高点,它通过最小能量路径将反应物和产物联系起来。由于过渡态存在时间极短,所以很难通过实验方法获得,应用线性同步转变二次同步转变法(LST/QT)来计算反应过渡态。反应能垒(Ea,kJ/mol)的计算如式(2)所示。
Ea=ETS-EIS
(2)
式(2)中,ETS和EIS分别为过渡态和初始态的能量, kJ/mol。
2 H2S及其分解基团在CuFe2O4表面的吸附机理
为研究H2S在CuFe2O4表面上的相互作用机理,首先对H2S分子及H2S分解产生的SH、S和H基团在CuFe2O4表面上的吸附进行计算。图1(b)中标注出了表面可能的活性位点,包括五配位FeO和CuO;3种三配位O:O1与2个八面体Cu和1个八面体Fe成键,O2与1个八面体Cu和2个八面体Fe成键,O3与1个八面体Cu、1个八面体Fe和1个四面体Fe成键。为便于后续分析,将H2S的分解基团SH、S和H基团在Fe、Cu、O1、O2、O3活性位的吸附分别命名为(1、2、3)A、(1、2、3)B、(1、2、3)C、(1、2、3)D、(1、2、3)E(如SH、S和H基团在Fe活性位的吸附分别命名为1A、2A和3A)。考虑了所有可能的吸附位点(Fe、Cu、O顶位,FeO—O、CuO—O桥位,以及穴位等),对H2S分子的吸附构型进行优化,结果表明H2S以物理吸附的形式吸附于表面,吸附能为-7.70~-21.15 kJ/mol。
2.1 SH基团的吸附
H2S分解可产生SH基团,对于SH基团在CuFe2O4(100)表面的吸附,考察了SH在CuFe2O4表面各个吸附位点上的所有可能吸附方式(SH平行、S向下、H向下)。图2为SH在CuFe2O4表面稳定的吸附构型和相应的吸附能。由图2可知,SH基团几乎均与表面成平行状态吸附。当SH基团吸附在Fe活性位时(1A),吸附能为-96.19 kJ/mol,S—Fe键长为0.2185 nm;当SH基团吸附在Cu活性位时(1B),吸附能为-58.79 kJ/mol,S—Cu键长为0.2221 nm。对比1A和1B 2种吸附构型的吸附能和S—Cu及S—Fe的键长可知,SH基团更容易以S原子化学吸附在Fe原子顶位上。SH基团在O原子顶位的吸附强弱按照吸附能由大到小排序为:O1、O2、O3。当SH基团吸附在O1活性位时(1C),吸附能为-164.16 kJ/mol,S—O键长为0.1631 nm; SH基团在O3活性位的吸附能(1E)相比在O1和O2原子上的吸附能显著降低,表明O3活性远低于另外2种O原子,S—O键长达到了0.1698 nm,说明SH基团中S原子与表面的O3相互作用较弱。
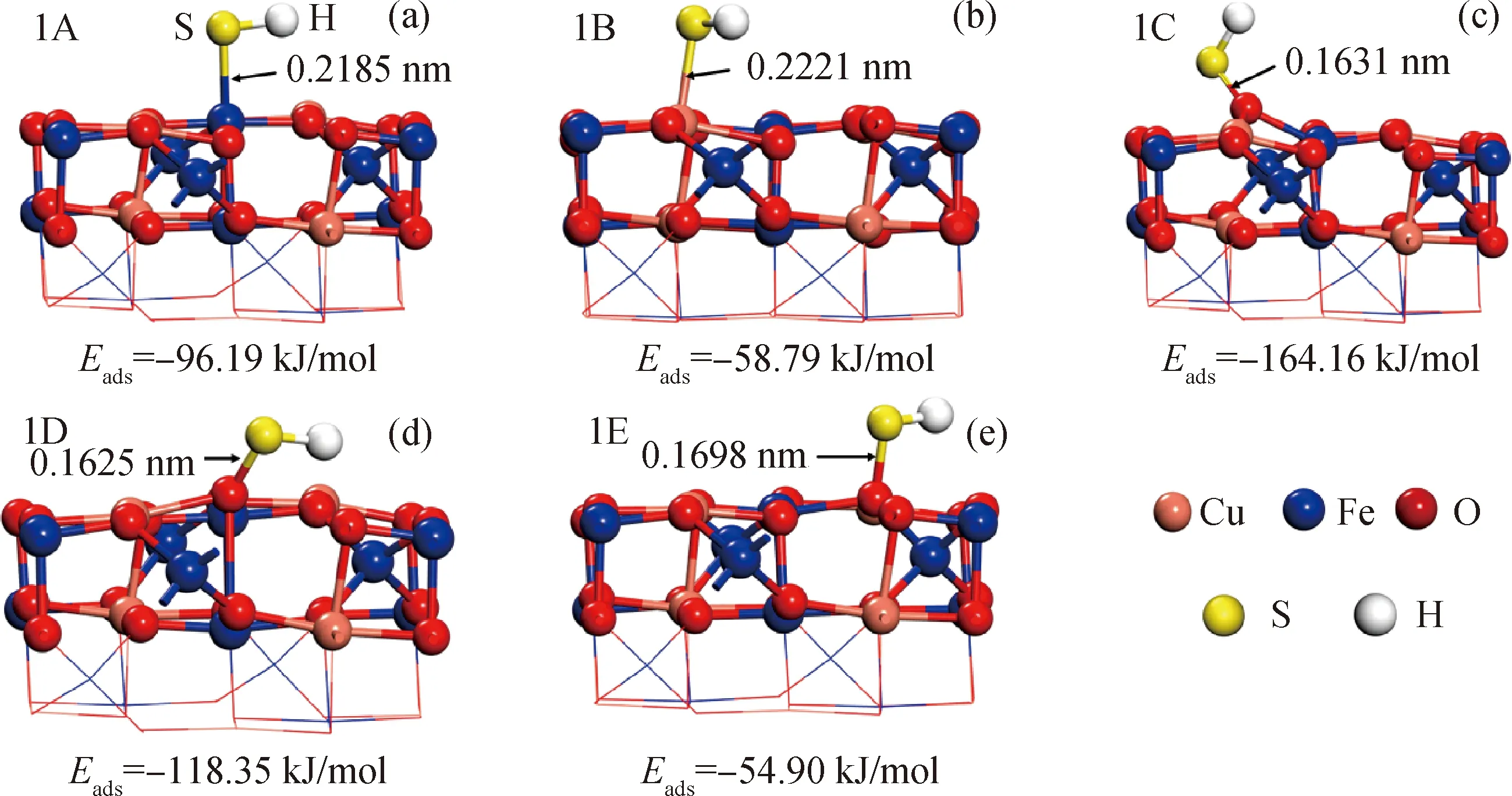
图2 SH在CuFe2O4表面的吸附构型Fig.2 Adsorption structures of SH on CuFe2O4 surface(a) 1A—SH adsorption on Fe site; (b) 1B—SH adsorption on Cu site; (c) 1C—SH adsorption on O1 site;(d) 1D—SH adsorption on O2 site; (e) 1E—SH adsorption on O3 site
2.2 S原子的吸附
S原子在CuFe2O4表面上的吸附构型及吸附能如图3所示。从图3可以看出,S原子在金属顶位上均可以稳定吸附。在构型2A中,S原子稳定吸附在Fe原子顶位,对应构型的吸附能为-147.44 kJ/mol,S—Fe键长为0.2046 nm;在构型2B中,S原子顶位吸附在Cu原子顶位,吸附能为-74.37 kJ/mol,S—Cu键长为0.2189 nm。从吸附能和键长可以看出,相比Cu顶位,S原子更倾向于吸附在Fe顶位上;在2C中,当S原子吸附在O1顶位时,对应的吸附能为-240.91 kJ/mol,是很强烈的化学吸附作用,S—O键长为0.1515 nm,载氧体表面结构发生明显变化;在构型2D中,S原子吸附在O2顶位时,其吸附能为-228.03 kJ/mol;在2E中,S原子吸附在O3原子顶位上,对应的吸附能为-160.65 kJ/mol,S—O键长为0.1675 nm,在表面3种不同的O原子活性位上,S均很容易实现稳定吸附,且吸附能均高于在金属原子活性位点的吸附,这可能是由于在O原子上吸附的构型表面重构造成的[32]。综上所述,S元素在CuFe2O4表面的吸附作用较强,很容易沉积在表面上,对CuFe2O4载氧体的反应性能造成不利影响。
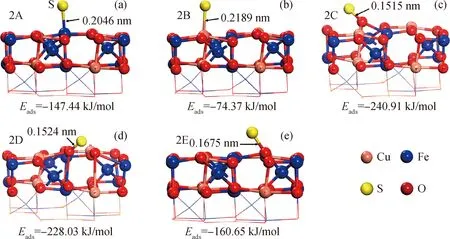
图3 S在CuFe2O4表面的吸附构型Fig.3 Adsorption structures of S on CuFe2O4 surface(a) 2A—S adsorption on Fe site; (b) 2B—S adsorption on Cu site; (c) 2C—S adsorption on O1 site;(d) 2D—S adsorption on O2 site; (e) 2E—S adsorption on O3 site
2.3 H原子的吸附
对于H原子在CuFe2O4表面的吸附,计算了H在FeO、CuO、O1、O2和O3顶位的吸附情况,相关的结构和吸附能量如图4所示。H在不同吸附位点的吸附能由大到小顺序为:O1、O2、O3、CuO、FeO。在构型3A中,吸附能为-152.88 kJ/mol,H—Fe键长为0.1494 nm;在构型3B中,吸附能为-199.37 kJ/mol,其吸附强度高于前者,说明H原子更倾向于吸附在表面上的Cu活性位点。在构型3C、3D和3E中,H原子在3种O活性位上的吸附能分别为-374.12、-348.48和-297.60 kJ/mol,H—O键长度范围在0.0982~0.0987 nm。由于3个 O原子位置的活性存在明显区别,因此导致三者的吸附能差别较大。其中H原子在O1原子位上的吸附活性最高,因此H原子在CuFe2O4表面上仍倾向于吸附在O1原子位上。
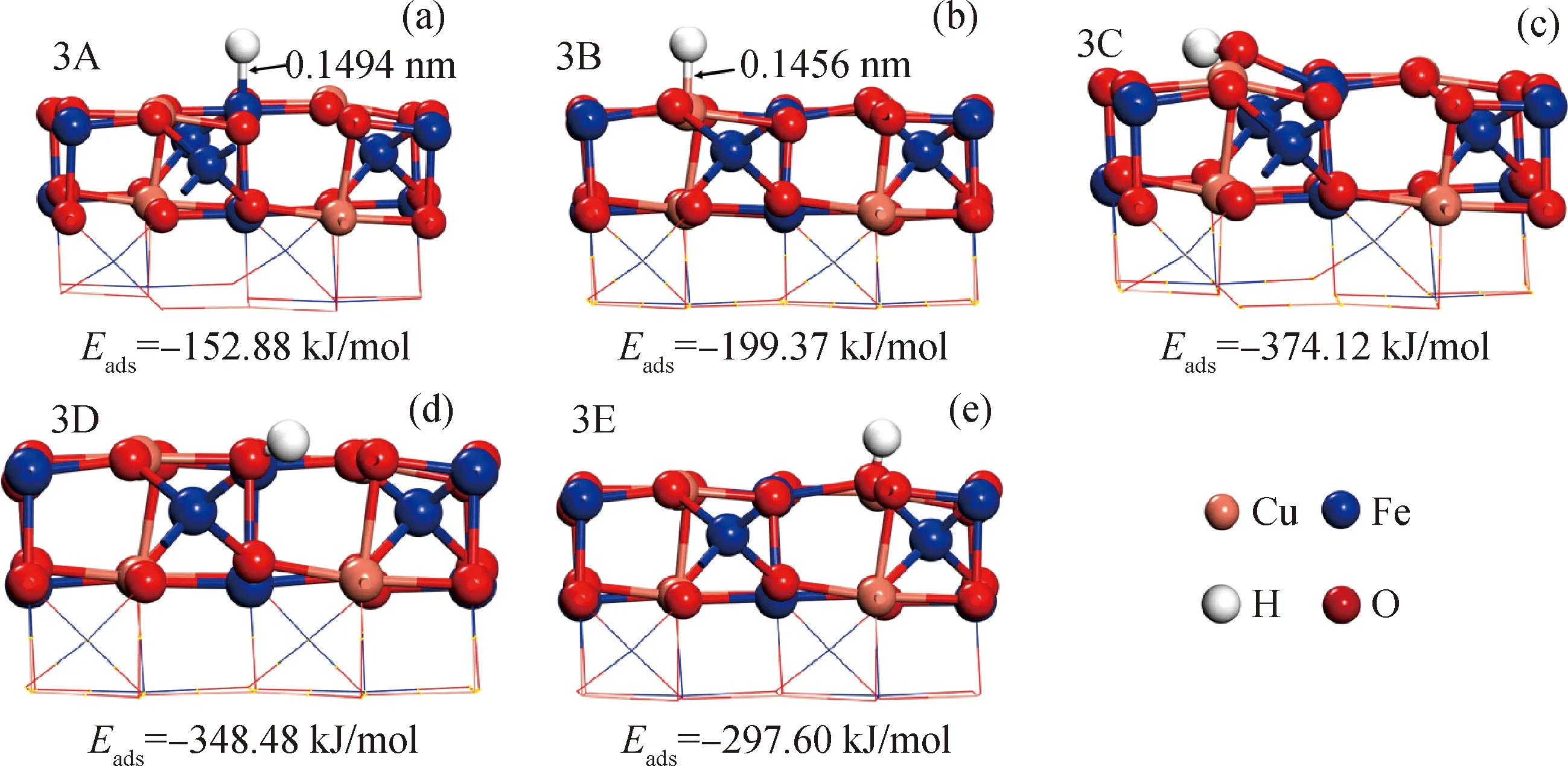
图4 H在CuFe2O4表面的吸附构型Fig.4 Adsorption structures of H on CuFe2O4 surface(a) 3A—H adsorption on Fe site; (b) 3B—H adsorption on Cu site; (c) 3C—H adsorption on O1 site;(d) 3D—H adsorption on O2 site; (e) 3E—H adsorption on O3 site
3 H2S与CuFe2O4表面的反应机理
通过上述对H2S分子及其解离基团吸附行为的分析,确定了不同基团的吸附活性位点及中间体结构。在此基础上进一步对H2S与CuFe2O4之间的反应机理进行计算。笔者通过基元反应步骤的过渡态搜索,比较不同反应路径的能垒[33],确定了H2S与CuFe2O4相互作用的主要反应路径,提出了CuFe2O4上H2S涉及的基元反应步骤,如式(3)~(6)所示。其中,*表示吸附在未占用的活性位点(如:SH*和S*分别表示吸附在未占用活性位点上的SH和S基团)。
H2S+*=H2S*
(3)
H2S*+*=SH*+H*
(4)
SH*+*=S*+H*
(5)
H*+OH*=H2O*
(6)
H2S与CuFe2O4之间的反应主要有2条路径。第一个反应路径(P1)是H2S分子首先在CuFe2O4表面的Fe顶位上吸附,进而发生两步脱氢反应,生成H2O脱离载氧体表面,载氧体表面的S基团发生迁移并填入氧空位形成硫化表面。第二个反应路径(P2)是H2S分子在表面的Cu顶位上发生两步脱氢反应,生成H2O,载氧体表面的S基团发生迁移并填入氧空位形成硫化表面。对这2种氧化反应路径进行了过渡态搜索,微观结构及势能变化如图5所示。
由图5可见,在P1路径中,H2S分子首先吸附在表面Fe原子上(IS1)。该吸附步骤放热13.46 kJ/mol,随后,吸附的H2S经历过渡态TS1发生第一步脱氢反应解离为SH基团和H原子(H2S*+*→SH*+H*)。解离后的H原子与表面的O1原子配位形成羟基(IM1),其对应的吸附能为-443.52 kJ/mol。CuFe2O4(100)表面H2S发生的第一步脱氢反应的反应能垒为99.03 kJ/mol、反应热为430.06 kJ/mol(即中间体IM1与初始态IS1体系总能量之差,-443.52-(-13.46)=-430.06)。
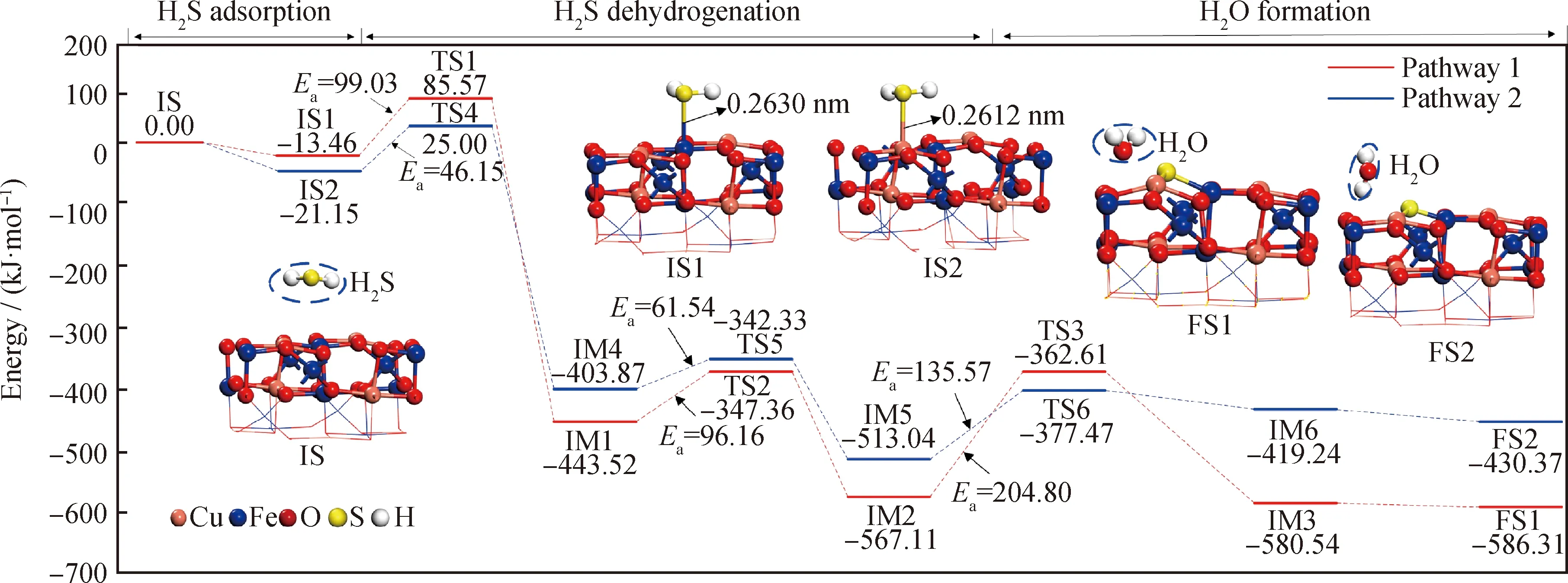
图5 H2S在CuFe2O4(100) 表面相互作用的反应路径Fig.5 The reaction pathway of H2S reaction on CuFe2O4(100) surface
在H2S的第二步脱氢步骤(SH*+*→S*+H*)中,IM1中间体经过渡态TS2将H原子从吸附在Fe顶位的SH基团上脱离形成羟基,S基团吸附在Fe顶位上(IM2),吸附能为-567.11 kJ/mol。TS2中S—H键长(0.2351 nm)大于IM1中S—H键长(0.2202 nm)和IM2中S—H键长(0.2038 nm),表明在第二步脱氢过程中,SH基团的活性先得到了提高,随后继续反应直到形成稳定的S*+H*+H*吸附结构。该脱氢步骤需克服96.16 kJ/mol的反应能垒,伴随着放出123.59 kJ/mol的热量(即中间体IM2与中间体IM1体系总能量之差,-567.11-(-443.52)=-123.59)。
H原子进而迁移到之前形成的羟基结构上,经历包含H2O分子前驱体结构的过渡态TS3,H2O分子从表面脱附,形成包含H2O前驱体的IM3,此步骤需克服的反应能垒约204.80 kJ/mol,释放出13.43 kJ/mol的热量(即中间体IM3与中间体IM2的体系总能量之差,-580.54-(-567.11)=-13.43)。随着反应的进行,载氧体表面的S基团发生迁移并填入氧空位(FS1),并伴随着释放5.77 kJ/mol的热量。在三步反应中,H2O的生成所需克服的反应能垒最高,为速控步骤。
在P2路径中,H2S分子吸附在CuFe2O4表面的Cu原子位上(IS2)。吸附后的H2S*进行第一步脱氢反应,经历TS4形成SH*+H*吸附结构(IM4),其中SH基团吸附在表面CuO原子上,其吸附能为 -403.87 kJ/mol,该过程需克服 46.15 kJ/mol的反应能垒,明显小于围绕表面Fe原子发生的脱氢反应所需克服的能垒(IS1→TS1→IM1,Ea=99.03 kJ/mol),这表明在反应过程中,CuFe2O4中Cu原子与S原子的反应活性高于Fe原子,会优先与H2S中S原子结合进行反应,根据文献[21,34-35]报道:在燃煤与CuFe2O4的化学链燃烧过程中,CuFe2O4中Cu组分不仅先于Fe组分被还原,而且Cu组分倾向与S元素反应生成Cu2S。在煤与CuFe2O4反应生成的固体硫化物中,Cu2S质量分数最高,达到了77.16%。这一结论从实验方面验证了该计算结果的有效性。
随后,SH基团发生第二步脱氢反应(SH*+*→S*+H*),IM4通过过渡态TS5将H原子从吸附在Cu顶位的SH基团上剥离形成羟基,S基团吸附在Cu活性位点上(IM5),对应的吸附能为-513.04 kJ/mol。该脱氢步骤需克服 61.54 kJ/mol 的反应能垒,并释放出约109.17 kJ/mol的热量(即中间体IM5与中间体IM4的体系总能量之差,-513.04-(-403.87)=-109.17)。经过比较,发现围绕Cu原子发生第二步脱氢反应的反应能垒(61.54 kJ/mol)明显小于围绕Fe原子的能垒(IM1→TS2→IM2,Ea=96.16 kJ/mol),这表明H2S在Cu原子顶位发生的第二步脱氢反应也是明显强于其在Fe原子顶位上的,这说明表面Cu位点的反应活性相较Fe位点的更强。
随着反应的进行,H原子移动到附近的羟基上,经历包含H2O分子前驱体的过渡态结构(TS6),形成包含H2O前驱体的中间体产物(IM6),在这个过程中需克服135.57 kJ/mol的反应能垒,并吸收93.80 kJ/mol的热量(即中间体IM6与中间体IM5的体系总能量之差,-419.24-(-513.04)=93.80)。经过比较,发现围绕Cu原子发生的形成H2O过程的反应能垒(135.57 kJ/mol)明显小于围绕Fe原子的能垒(IM5→TS6→IM6,Ea=204.80 kJ/mol),这是因为O1原子的反应活性最高,而O1原子与2个Cu原子和1个Fe原子配位,因此围绕Cu原子形成H2O的P2路径更容易发生,文献[29]所述CuFe2O4表面与2个Cu原子和1个Fe原子配位的O原子活性最高,CO分子与该原子相互作用十分强烈,可使O1原子脱离表面而与CO直接形成CO2分子,该研究结果从侧面佐证了这一点。最终,吸附在Cu原子顶位的S基团发生迁移并填补氧空位(FS2),并释放出11.13 kJ/mol的热量(即终态FS2与中间体IM6的体系总能量之差,-430.37-(-419.24)=-11.13)。在P2反应路径中,H2O的生成也为速控步骤。
在2条反应路径中,对于围绕Cu活性位点进行反应的P2路径,H2S两步脱氢反应及H2O生成的反应能垒均比围绕Fe活性位点的更低,因此认为P2路径是H2S与CuFe2O4表面反应的主要反应路径。
4 结 论
基于密度泛函理论和周期性结构模型研究了H2S在CuFe2O4载氧体表面的相互作用机理,得出以下结论:
(1)H2S分子解离的SH、S和H基团倾向于吸附在CuFe2O4表面的O1活性位点上,相应的吸附能分别为-164.16、-240.91和-374.12 kJ/mol。
(2)H2S与CuFe2O4表面反应主要包括3个基元步骤:H2S分子吸附、H2S脱氢和H2O分子形成。首先H2S吸附在CuFe2O4表面进而发生两步脱氢反应,随着反应的进行,产生的H2O分子从表面脱附,载氧体表面的S基团发生迁移并填入氧空位形成硫化表面。H2O分子的形成需克服135.57 kJ/mol的反应能垒,为速控步骤。
(3)在H2S与CuFe2O4表面的反应过程中,围绕Cu活性位点进行的H2S两步脱氢反应及H2O生成的反应能垒均比围绕Fe活性位点的能垒更低,因此围绕Cu原子进行的反应路径是主要路径。
