基于蛋白质结构域特异性的关键蛋白质识别算法*
2020-03-04杨増光
杨増光
(南京理工大学 南京 210094)
1 引言
众所周知,蛋白质(Protein)在细胞的组成和生物体的生命活动中扮演着极其重要的作用。但不同类型的蛋白质对生物体的重要程度不尽相同,其中那些缺失后会导致生物体病变甚至死亡的蛋白质被称为关键蛋白质(essential proteins),其余的则被称为非关键蛋白质(non-essential proteins)[1~3]。
研究表明,关键蛋白质的识别对于我们了解细胞的生长调控过程,研究生物进化的相关机制,以及根据关键蛋白质进行药物设计、药物标靶鉴定和疾病治疗等方面具有着不可忽视的现实意义[4]。
在生物学领域中,识别关键蛋白质通常是采用生物医学实验的方式进行的,这类方法虽然准确,但是成本高、效率低,无法适用于日益增长的蛋白质数据。随着高通量技术的发展,越来越多的蛋白质相互作用数据被获取,这让我们能够从网络水平上识别关键蛋白质。
目前,越来越多的研究人员将图论、复杂网络等相关知识应用到蛋白质网络中,并提出多种有效的方法来识别关键蛋白质,其中常用的有8种具有代表性的算法:DC[5]、BC[6]、CC[7]、SC[8]、EC[9]、IC[10]、LAC[11]、NC[12]。这些算法虽然能够有效地识别出关键蛋白质,但是由于这类算法容易受到网络中假阴性和假阳性数据的影响且忽略了蛋白质网络蕴含的生物信息,因而它们的识别精度不高。
本文,我们提出一种基于蛋白质结构域特异性的关键蛋白质识别算法Do-ECC,通过融合蛋白质网络的拓扑信息和生物信息,能够有效提高关键蛋白质的识别准确度。
2 边聚集系数
为充分利用蛋白质网络的拓扑信息,首先需要寻找一个有效的拓扑特征。研究表明,关键蛋白质更可能和关键蛋白质相连,并且成簇出现,而非关键蛋白质则表现稀疏,即关键蛋白质在网络中所处的位置相比于非关键蛋白质拥有更高的连通度和模块化程度[11~12]。基于此,越来越多的研究人员开始使用边聚集系数作为描述蛋白质网络的拓扑特征来开展自己的研究,实验结果也表明,这一特征确实能够更全面、更准确地描述蛋白质网络的拓扑信息。
对网络中的任一条边,边聚集系数被定义为该边在网络中实际参与构成的三角形个数与该边最多可能参与构成的三角形个数之比。如对于边E(u,v),其边聚集系数可表示为
其中zu,v表示网络中该边实际参与构成的三角形的个数,ku和kv分别表示节点u和v的度,则表示该边最多可能参与构成的三角形的个数。不难看出,边聚集系数的取值介于0~1之间。对于任一条边,其边聚集系数越大,表明其参与网络模块结构的比重越多,在网络中所处位置的聚集程度也越高。
3 蛋白质结构域特异性
大多数蛋白质通常是由一个或者多个功能区域组成,这些区域一般被称为蛋白质结构域(Protein Domain),是蛋白质结构和功能的基本单位。而在自然界中,复杂的蛋白质分子则是由这些结构域通过不同的组合和重排形成的。研究表明,那些在生物体中出现频率较少的结构域对于生物体具有更加关键的作用;另一方面,包含较多结构域的蛋白质分子,通常执行更多的生物功能,对正常的生命活动更加重要,也更有可能是关键蛋白质[13]。
3.1 TF-IDF算法
在信息检索、文本分类等相关领域,TF-IDF(Term Frequency-Inverse Document Frequency),即“词频-逆文本频率”,是一种常用的加权技术,用以评估一个单词对于文本和语料库的区分能力与重要程度。
其中TF(Term Frequency),即词频,是指一个单词在一个文本中出现的频率,通常表示为这个单词在文本中出现的次数和该文本包含的单词的总数之比,即:

其中,ni,j表示单词i文本 j中出现的次数,k表示文本 j包含的单词类型的数目,则表示该文本包含的单词的总数。
IDF(Inverse Document Frequency),即逆文本频率的概念,对于一个单词,它是指整个语料库中包含该单词的文本的数量,通常表示为先计算语料库的文本总数和包含该单词的文本数之比,然后取对数,即:
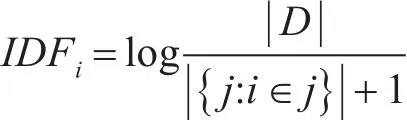
而TF-IDF就是通过结合两者,用来评估一个单词对于文本和语料库的区分能力与重要程度,通常表示为

根据上述定义,可以发现,一个单词的重要性和区分能力随着它在文本中出现的次数成正比增加,但同时会随着它在整个语料库中出现的频率成反比下降。
3.2 结构域特异性
借鉴TF-IDF算法的思想,我们对蛋白质结构域进行重新审视。如果将每种类型的结构域都当作一个单词,那么每条蛋白质就相当于一个文本文件,而整个生物体包含的所有蛋白质就组成了一个语料库。如图1所示,如果将PF00270、PF00271等几种结构域视作一种单词,则蛋白质YER172C、YBL084C、YDL126C的“文本”组成可以表示如图1所示。
根据IDF的定义,本文提出了IPF(Inverse Protein Frequency)的概念,来描述蛋白质结构域的特异性,即由生物体包含的蛋白质总数除以包含该结构域的蛋白质数目,再将得到的商数取对数,如下所示:

图1 蛋白质的结构域组成示意图

同理,根据TF的定义,本文提出DF(Domain Frequency)的概念,指一个结构域在一个蛋白质分子中出现的频率,表示为这个结构域在特定蛋白质分子中出现的次数和该蛋白质包含的结构域的总数之比,即:
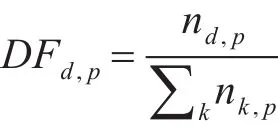
其中,nd,p表示蛋白质结构域d在蛋白质分子 p中出现的频次,k表示蛋白质分子 p包含的结构域种类数,则表示蛋白质分子 p包含的结构域的总数。
3.3 蛋白质的结构域特异性得分
参照TF-IDF的定义,本文给出DF-IPF的概念。对于结构域d,其特异性为IPFd,在蛋白质 p中出现的频率为DFd,p,则它对应的DF-IPF值可以表示如下:

它可以用于描述一个结构域d对蛋白质 p的重要程度,也可以用于度量蛋白质 p基于结构域d获得的特异性得分。而一个蛋白质可能包含多种类型的结构域,则其总的结构域特异性得分可以表示为

其中,k表示蛋白质 p包含的结构域的种类。
4 Do-ECC算法
如上所述,边聚集系数能够描述蛋白质网络的拓扑信息,蛋白质的结构域特异性得分则反映了蛋白质网络蕴含的生物信息。本文,我们通过融合这两种特征,提出一种新的关键蛋白质识别算法Do-ECC。
为方便介绍,首先对蛋白质网络进行建模,将其表示成一个无向图G(V ,E ),如对于存在相互作用的两个蛋白质分子,可以将这两个蛋白质分别表示为节点u和v,而将它们间的相互作用表示边E(u ,v )。
根据上述定义,对于相互作用E(u ,v) ,其边聚集系数可以表示为ECC(u ,v)。为能够和结构域信息进行融合,需对ECC(u ,v)进行归一化处理,表示为

其中,ECCMAX和ECCMIN分别表示所有相互作用的边聚集系数的最大值和最小值。
对于蛋白质节点u,它的结构域特异性得分可以表示为Spec()u,同样需要进行归一化处理,表示为

其中,SpecMAX和SpecMIN分别表示所有蛋白质分子的结构域特异性得分的最大值和最小值。同理,对于蛋白质节点v,其归一化处理后的结构域特异性得分可以表示为SpecNORM()v。
研究表明,蛋白质的关键性和蛋白质分子间的相互作用存在密切关系,因此我们可以通过相互作用的两个蛋白质的结构域特异性计算出这条相互作用的特异性。如对相互作用E(u ,v) ,其结构域特异性得分取决于它对应的两个蛋白质分子u和v,可以表示为

Do-ECC算法就是通过结合两者来评估蛋白质的关键性,如对蛋白质节点u,其关键性得分可以表示为

其中Nv是节点u的邻居节点的集合,v是节点u的邻居节点且v∈Nv。蛋白质节点的关键性得分越高,越可能是关键蛋白质。
5 实验
5.1 实验数据
1)蛋白质相互作用数据
鉴于酵母的蛋白质相互作用数据的相对完备性,本实验选择酵母作为研究对象。所用的蛋白质相互作用数据是从DIP数据库[14]下载获得,采用的数据集版本是2017年2月5日更新的酿酒酵母的全部蛋白质相互作用数据集。原始数据集中包含22977条蛋白质相互作用,去除自连接和重复的相互作用后,共提取出22620条相互作用,包含5126个蛋白质分子。
2)蛋白质结构域数据
本实验所用到的蛋白质结构域数据是从PFAM数据库[15]中下载获得的,采用的数据集版本是于2017年3月份更新的Pfam 31.0。因为在PFAM数据库中,有两种不同质量水平的结构域序列数据:Pfam-A系列和Pfam-B系列。其中,Pfam-A系列的数据质量水平较高,而Pfam-B系列的数据未经注释过且质量水平也较低,因此,本实验仅仅提取酵母的Pfam-A系列的结构域序列数据。在实验中,我们通过在PFAM数据库中下载获取到swisspfam.gz文件,经过预处理后,提取出具有已知的结构域信息的蛋白质共4174个,包含了2829种结构域,而剩余的952个蛋白质则认为没有已知的结构域信息。
3)已知的关键蛋白质和非关键蛋白质
通过实验得到的候选关键蛋白质需要和目前已知的关键蛋白质数据进行比对,进而分析实验方法的有效性和准确率。本实验所选用的已知关键蛋白质数据是通过整合数据库 SGD[16]、DEG[17]和SGDP[18]中的酵母的关键蛋白质信息数据得来。最后整合得到的酿酒酵母的关键蛋白质1299个,非关键蛋白质4982个。将从DIP数据库中获取的酵母的5126个蛋白质分子与已知关键蛋白质和非关键蛋白质数据对比后,我们发现可以将5126个蛋白质分子分为3类:关键蛋白质、非关键蛋白质和关键性未知的蛋白质,其中含有关键蛋白质1159个,非关键蛋白质3612个,关键性未知的蛋白质355个。在实验过程中,我们将关键性未知的蛋白质归为非关键蛋白质一类。
5.2 评价指标
通常来讲可以将关键蛋白质的识别问题当作非监督的分类问题,然后采用统计学中常用的“排序-筛选”的方法对不同的关键蛋白质识别算法的实验结果进行比较和分析[19]。针对本实验,“排序-筛选”方法的具体过程如图2所示。
除此之外,为更加有效地对各个算法的实验结果进行评估,还可以使用6种常用的测量指标,包括敏感度(Sensitivity,SN)、特异性(Specificity,SP)、F-测度(F-measure)、正确率(Accuracy,ACC)、阳性预测值(Positive Predictive Value,PPV)和阴性预测值(Negative Predictive Value,NPV)。在详细分析这几种指标之前,首先需要了解表1中介绍的几个概念。

图2 排序-筛选的流程

表1 相关概念简介
基于表1中介绍的四个基本概念,这6种常用的检测指标定义如下:

5.3 实验结果与分析
按照“排序-筛选”的方法,我们首先计算出5126个蛋白质节点在上述各个算法下的测度参数并根据测度值按降序排序,然后分别挑选前1%、5%、10%、15%、20%以及25%的部分作为候选的关键蛋白质,最后将其和已知的键蛋白质数据进行对比,得出各个算法识别出的正确的关键蛋白质数目,如表2所示。
由表2展示的实验结果,可以发现,Do-ECC算法识别出的正确的关键蛋白质数目在各个范围内均显著多于其他8种算法。
为更加细致地比较各个算法识别关键蛋白质的效果,进一步使用SN、SP、F、ACC、PPV和NPV对它们的实验结果进行评估比较,如表3所示。
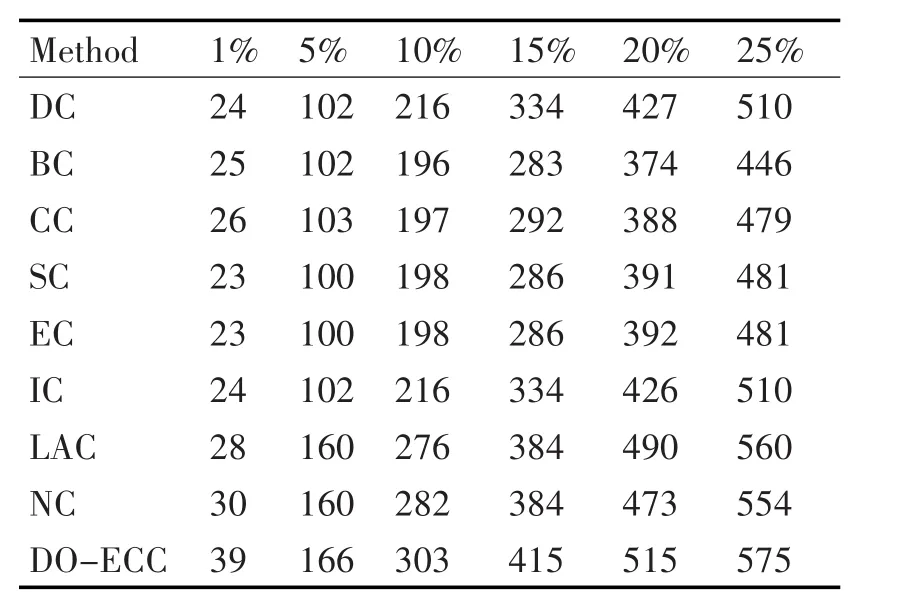
表2 九种算法识别出的正确的关键蛋白质数目

表3 九种算法在6种常用检验指标下的实验结果
由表3不难看出,Do-ECC算法在SN、SP等6种指标下的得分均高于其他8种基于蛋白质网络拓扑特征的算法。
6 结语
本文使用边聚集系数刻画蛋白质网络的拓扑特征,并借鉴TF-IDF算法的思想,提出蛋白质结构域特异性的概念,然后融合蛋白质网络的拓扑信息和生物信息,提出一种基于蛋白质结构域特异性的关键蛋白质识别算法Do-ECC,最后通过实验验证了所提蛋白质结构域特异性和Do-ECC的有效性。
