基于Illumina MiSeq测序技术不同地区辣椒酱细菌多样性分析
2020-03-02宁明赵馨馨董蕴倪慧单春会张振东郭壮
宁明,赵馨馨,董蕴,倪慧,单春会,张振东,郭壮,3*
(1.湖北文理学院 食品科学技术学院 鄂西北传统发酵食品研究所,湖北 襄阳 441053; 2.石河子大学 食品学院,新疆 石河子 832000;3.恩施市公共 检验检测中心,湖北 恩施 445000)
辣椒酱因微生物的发酵作用,兼具营养丰富、味美色艳等特点广受消费者喜爱,是我国传统发酵食品[1]。徐廷弼等[2]采用传统培养和16S rDNA测序的方法对存在产气现象的辣椒酱生产设备和空气中的细菌菌群种类和含量进行了解析,结果显示:乳杆菌属和芽孢杆菌属在辣椒酱中占明显优势。丁文等[3]分别采用16S rDNA和ITS序列鉴定分离的细菌和真菌菌株,并运用PCR-DGGE法分析贮藏终点辣椒酱中细菌群落结构,发现贮藏终点优势细菌为乳酸菌和蜡样芽孢杆菌。传统辣椒酱中具有相对较高的微生物多样性,不仅蕴含具有潜在益生功能及影响产品品质形成的菌株,亦可能存在食源性致病菌[4]。越来越多的研究表明不同地区同一类发酵食品中微生物多样性可能存在一定的差异[5],而关于咸丰地区辣椒酱中细菌群落结构多样性的研究鲜见报道。
与其他二代高通量测序技术相比,Illumina MiSeq平台采用宏基因组学的研究策略,不仅大大降低了成本,而且增加了每个样品的测序深度,兼具测序量大和精确度高等特点[6],目前在发酵食品和动物肠道微生物多样性解析领域有着广泛的应用[7,8],这为全面反映辣椒酱中细菌微生物群落特点提供了新的研究方法。目前对于辣椒酱的研究主要集中于品质分析或特定性能菌株的筛选等制作环境相对开放的传统辣椒酱中[9,10],而对于辣椒酱中微生物群落结构的分析鲜有报道。
本研究以采集的辣椒酱为研究对象,采用Illumina MiSeq高通量测序技术对其微生物多样性进行深入解析,同时对不同地区辣椒酱的群落结构进行比较分析。在对辣椒酱中细菌群落进行全面解析的同时,探讨挖掘核心细菌类群,以期为功能性菌种资源的开发及产品的产业化生产提供理论支持。
1 材料与方法
1.1 材料与主要试剂
辣椒酱:从湖北省恩施土家族苗族自治州咸丰县农贸市场采集10个辣椒酱样品,分别命名为XFLJ1~XFLJ10,每个样品200 g左右,制作时间在15~20 d。
QIAGEN DNeasy mericon Food Kit提取试剂盒:德国QIAGEN公司;dNTPs Mix、Fast Pfu Buffer、5×Trans StartTM、Fast Pfu Fly DNA Polymerase:宝生物工程(大连)有限公司;338F/806R正向及反向引物:武汉天一辉远生物科技有限公司。
1.2 仪器与设备
5810R台式高速冷冻离心机 德国Eppendorf公司;VeritiTM96孔梯度PCR扩增仪 美国AB公司;FluorChem FC3型化学发光凝胶成像系统 美国ProteinSimple公司;DYY-12电泳仪 北京市六一仪器厂;Illumina MiSeq高通量测序平台 美国Illumina公司;R930机架式服务器 美国DELL公司。
1.3 实验方法
1.3.1 DNA提取
称取10 g辣椒酱样品加入蒸馏水40 mL后于300 r/min条件下离心10 min取上清液,再以10000 r/min离心10 min后保留沉淀,采用QIAGEN DNeasy mericon Food Kit试剂盒提取微生物宏基因组DNA。
1.3.2 PCR扩增及MiSeq高通量测序
以纯化后的DNA为模板,采用包含“5′-ACTCCTACGGGAG-GCAGCA-3′”序列的正向引物和“5′-GGAC-TACHVGGGTWTCTAAT-3′”的反向引物扩增16S rDNA V4~V5区进行高通量测序分析。
聚合酶链式反应(polymerase chain reaction,PCR)体系[11]:扩增体系为20 μL,10 ng模板DNA,0.8 μL 5 μmol/L正反向引物,5 U/μL DNA聚合酶0.4 μL,2.5 mmol/L dNTPs mix 2 μL,5×PCR缓冲液4 μL,使用超纯水补充剩余体系。PCR反应条件为95 ℃ 3 min,然后执行95 ℃ 30 s,55 ℃ 30 s,72 ℃ 45 s,共35 个循环,完成72 ℃ 10 min后于4 ℃保存。经琼脂糖凝胶电泳检测合格后的PCR产物寄往上海美吉生物医药科技有限公司完成测序。
1.3.3 生物信息学分析
采用QIIME软件平台调用UPARSE进行两步UCLUST法聚类[12],继而应用Chimera Slayer去除嵌合体序列[13],从而得到优化序列用于操作分类单位(operational taxonomic unit,OTU)分析,整合RDP和Greengenes两个数据库确定细菌OTU代表性序列的微生物分类水平并统计每个样本的群落组成[14,15],同时一方面对辣椒酱细菌微生物的超1(Chao1)和香农(Shannon)指数等α多样性指标进行计算[16],另一方面使用基于分类操作单元加权UniFrac距离[17]的主成分分析法(principal component analysis,PCA)和非加权组平均法(unweighted pair group method using arithmetic average,UPGMA)聚类进行β多样性等一系列群落结构和组间差异的统计学和可视化分析。
1.3.4 数据下载及上传
本研究首先从MG-RAST数据库(http://www.mg-rast.org)中下载当阳地区辣椒酱细菌相关序列(ID号为mgp82587)。咸丰地区10 个辣椒酱样品MiSeq高通量测序数据均已提交至MG-RAST数据库(mgp89819)。
1.4 多元统计学分析及图表绘制
应用方差分析(multivariate analysis of variance,MANOVA)手段解析2类地区辣椒酱细菌群落结构并采用Mann-Whiney检验对其显著性进行分析;通过LDA算法甄别与细菌群落结构显著差异相关的关键类群并通过各样品之间的欧式距离矩阵计算组间差异。系统发育树由Mega 6.0软件绘制,其他图均在Origin 8.6软件中完成绘制。
2 结果与分析
2.1 样品16S rRNA基因序列测序深度及质量评估
经质控合格后,本研究共获得379918条高质量序列,平均每个样品序列为37992条。序列按照97%相似性进行操作分类单元划分后,共得到25453个OTU。辣椒酱样品16S rRNA测序结果及各分类水平数量见表1。

表1 辣椒酱样品测序结果及各分类地位数量Table 1 Sequencing results and number at different taxonomical levels of chili sauce samples
注:“*”表示细菌超1指数和香农指数均在测序量为22010条序列时计算所得。
由表1可知,在10个辣椒酱样品中XFLJ9的超1指数最大,而样品XFLJ1的香农指数最大,这说明XFLJ9样品具有最高的细菌物种多样性,而XFLJ1样品细菌物种丰度最大。在OTU划分的基础上,所有序列划分为133个门、316个纲、488个目、851个科和1342个属,其中有0.09%和7.18%的序列不能鉴定到门和属水平。
2.2 基于各分类学地位辣椒酱中细菌相对含量的组成分析
为得到每个OTU对应的物种分类信息,本研究在序列丰富度和多样性分析的基础上,采用97%相似水平的OTU代表序列进行分类学分析,并在门水平统计每个样品的群落组成。根据各个样品不同门的细菌所占比例作图,将相对丰度低于1.0%的门合并为其他,不同样品中细菌在分类门水平上的分布情况见图1。
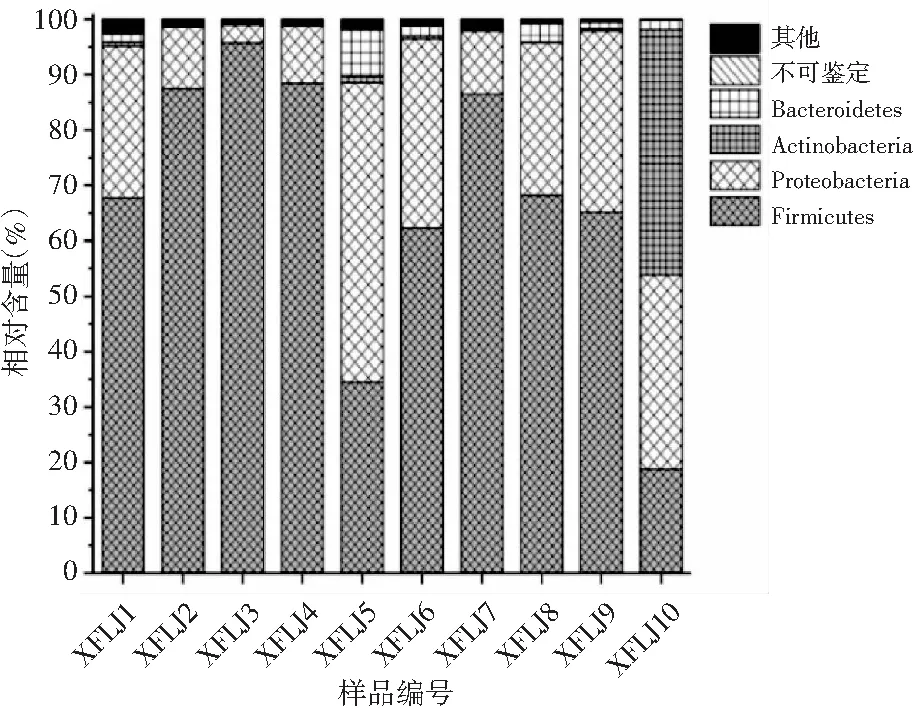
图1 细菌在门水平的组成及相对含量Fig.1 The composition and relative content of bacteria at the phylum level
由图1可知,在辣椒酱10个样品中(XFLJ1~XFLJ10)共鉴定出33个门,平均丰度大于1.0%的有4 个门,分别隶属于厚壁菌门(Firmicutes)67.46%、变形杆菌(Proteobacteria)24.71%、放线菌(Actinobacteria)4.75%和拟杆菌(Bacteroidetes)1.83%,其中仅有0.09%的序列不能鉴定到门水平。从门的水平上讲,细菌的种类大致相同,但丰度差别较大。
10个辣椒酱样品中细菌在分类属水平上的分布情况见图2。
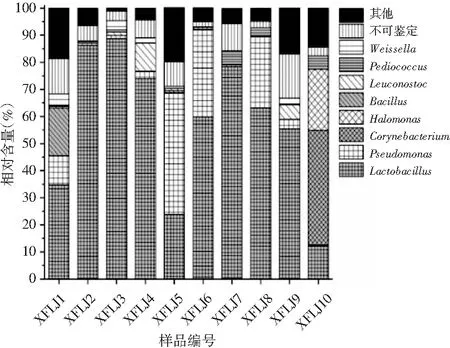
图2 细菌在属水平的组成及相对含量Fig.2 The composition and relative content of bacteria at the genus level
由图2可知,样品中相对含量大于1.0%的细菌属有8个,分别为隶属于厚壁菌门(Firmicutes)的乳酸杆菌(Lactobacillus,57.66%)、芽孢杆菌(Bacillus,12.25%)、明串珠菌(Leuconostoc,4.29%)、片球菌(Pediococcus,2.29%)和魏斯氏菌(Weissella,1.97%);隶属于变形杆菌(Proteobacteria)的假单胞菌(Pseudomonas,1.90%)、盐单胞菌(Halomonas,1.44%)和隶属于放线菌(Actinobacteria)的棒状杆菌(Corynebacterium,1.30%)。
结果显示:在属水平上,不同样品中所含细菌种类的多样性及其丰度存在较大差异。对于样品XFLJ3而言,乳杆菌(Lactobacillus)、明串珠菌(Leuconostoc)和魏斯氏菌(Weissella)是主要的菌属,其平均丰度在31.11%左右,未知属的丰度达到3.60%,其他种类的属的含量所占比例都较低。隶属于厚壁菌门(Firmicutes)的乳酸杆菌(Lactobacillus)和魏斯氏菌(Weissella)能够产生多种风味物质,改善发酵食品的风味缺点,使其兼具益生和味美的双重功能[18],其可作为丰富和完善乳酸菌资源库和基因库的菌种来源。相对而言,样品XFLJ10中物种多样性最为丰富,其中棒状杆菌(丰度为42.44%)和盐单胞菌(丰度为22.54%)是主要的微生物,其含量远高于辣椒酱中其他样品;乳酸杆菌(Lactobacillus)和片球菌(Pediococcus)的丰度分别为12.17%和4.87%。此外,在研究中发现辣椒酱XFLJ1中含有17.60%的芽孢杆菌(Bacillus),这可能与其具有较高浓度的食盐耐受性有关,同时芽孢杆菌亦作为影响浓香型大曲等发酵食品的关键微生物类群[19],对其风味品质的工艺优化具有积极的影响。本研究结果表明,辣椒酱中细菌的构成较为复杂,菌群的种类和数量也不尽相同。
2.3 基于多元统计学分析不同地区辣椒酱细菌群落结构
为确定影响辣椒酱中细菌群落结构的因素,根据前期对湖北省当阳地区辣椒酱的研究[20],本研究进一步在分类操作单元非加权欧式距离的PCA主坐标分析及UPGMA的基础上对16个辣椒酱(10个咸丰地区辣椒酱和6个当阳地区辣椒酱)样品的β多样性进行进一步的比较分析,基于OTU水平非加权UniFrac距离显著性检验的主坐标分析见图3。
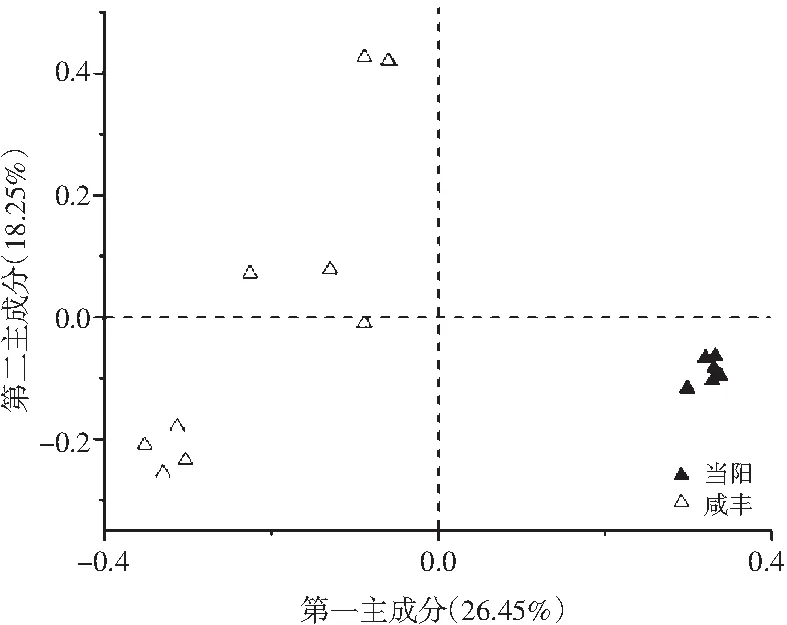
图3 基于非加权UniFrac距离的细菌主坐标分析Fig.3 Principal coordinate analysis of bacteria based on unweighted UniFrac distance
由图3可知,以权重最高的PC1(26.45%)和PC2(18.25%)两个主成分绘制PCA,当阳地区辣椒酱样品主要分布在第一象限,而咸丰地区辣椒酱样品分布在第二、第三象限。由此可知,虽然辣椒酱样品中存在大量的核心细菌菌群,但2类地区辣椒酱距离相差较远,这说明2个样本间可能存在较大的差异。为了能够更加清晰地反映样品菌群结构存在的差异,本研究进一步采用UPGMA显著性检验对2类辣椒酱样品间的差异性进行层次聚类分析,基于OTU的16个辣椒酱样本的聚类分析见图4。

图4 基于OTU水平非加权UniFrac距离的 UPGMA聚类分析Fig.4 UPGMA cluster analysis of OTU level based on unweighted UniFrac distance
由图4可知,用于比较的16个辣椒酱样品大致可分为3个大类,其中咸丰地区辣椒酱样品XFLJ1~XFLJ9和当阳地区辣椒酱样品DYLJ1~DYLJ6各为一类,而XFLJ10样品被聚为一类,这与主成分分析结果基本一致。整体从16个样品来看,相似度差别较大,这可能与地域差异性导致的辣椒酱核心类群的不同有关。结合图3可知,造成这种现象的原因可能是辣椒酱样品XFLJ10相较于其他9个样品来说,有相对含量远高于其他样品的盐单胞菌(Halomonas)和棒状杆菌(Corynebacterium)等优势细菌属的存在。
在非加权UniFrac水平上,分支的长度能够较好地反映群落的相对丰度差异,由图4结果还可知,咸丰地区辣椒酱样品各分支长度整体上要大于当阳地区辣椒酱样品各分支,为探究其显著差异性,本研究进一步使用欧氏距离对辣椒酱样品细菌群落结构组间差异进行解析,结果见图5。
由图5可知,通过采用欧式距离对当阳地区(0.522±0.025)和咸丰地区(0.659±0.0743)辣椒酱微生物群落结构组间差异进行分析的基础上,由Mann-Whiney检验得出2组样本间差异非常显著(p<0.01),说明咸丰地区细菌群落结构组间差异要显著高于当阳地区。

图5 基于非加权欧氏距离的细菌群落结构组间差异分析Fig.5 Differences in the bacterial community structure based on unweighted UniFrac distances
注:“**”表示差异非常显著,p<0.01。
2.4 基于分类水平的关键细菌类群的甄别
根据上述研究结果分析发现,咸丰地区和当阳地区的辣椒酱微生物群落结构差异极显著(p<0.01)。本研究以辣椒酱分组(咸丰地区/当阳地区)为起非监督作用的解释变量,以LDA分数(LDA Score)取对数(log10)之后相对值大于3的分类操作单元为响应变量,甄别了影响2类地区辣椒酱微生物群落结构在丰度上差异显著的关键细菌。

图6 LDA值分布柱状图Fig.6 The distribution histogram of LDA values
由图6可知,2组样品具有统计学差异,从宏观上描述了所有类群和总体之间的离散冗余程度,2类辣椒酱在该空间中具有最佳的可分离性。由此可见,12个主要细菌类群代表了2类辣椒酱微生物群落结构差异显著相关的关键细菌类群。隶属于Firmicutes(厚壁菌门)的Bacillaceae(芽孢杆菌)科和Bacillus;隶属于Actinobacteria(放线菌)的Actinomycetales,Brevibacteriaceae,Brevibacterium(短杆菌)和Kocuria;隶属于Proteobacteria(变形菌门)的Aeromonadaceae,Rhodobacterales,Rhodobacterceae及Massilia(位于图的左侧,即咸丰地区辣椒酱),这说明该10个类群在咸丰地区辣椒酱中的相对含量可能较高;隶属于Proteobacteria(变形菌门)的Ramlibacter和Hafniaceae(位于图的右侧,即当阳地区辣椒酱),这说明该2个细菌类群在当阳地区辣椒酱中的相对含量可能较高。整体上看,咸丰地区辣椒酱细菌群落结构种类多样性及丰度要显著高于当阳地区,这与图6的结果一致。
3 结论
MiSeq高通量测序结果表明,咸丰地区10个辣椒酱样品中的优势细菌类群主要是由隶属于厚壁菌门(Firmicutes)的乳酸杆菌(Lactobacillus)、芽孢杆菌(Bacillus)、明串珠菌(Leuconostoc)、片球菌(Pediococcus)和魏斯氏菌(Weissella);隶属于变形杆菌(Proteobacteria)的假单胞菌(Pseudomonas)和盐单胞菌(Halomonas)及隶属于放线菌(Actinobacteria)的棒状杆菌(Corynebacterium)构成。通过对比当阳地区辣椒酱细菌类群,对辣椒酱样品中细菌群落结构中关键细菌类群进行评价,发现咸丰地区辣椒酱细菌群落结构种类多样性及丰度要显著高于当阳地区辣椒酱。
