氢氘交换质谱技术在蛋白质和蛋白复合物结构研究中的应用进展
2020-02-28李惠琳
黄 静,李惠琳,2*
(1.中山大学 药学院,广东 广州 510006;2.中山大学 药学院,广东省手性分子与药物发现重点实验室,广东 广州 510006)
蛋白质(Protein)是生命活动的物质基础,调控生命体内几乎所有的生物学过程[1-2]。蛋白质在一级结构(氨基酸序列)的基础上折叠、盘曲成特定的空间结构而具有功能[3],其功能受结构动态变化[4-5]、配体(如小分子、蛋白质或DNA等)相互作用及翻译后修饰(Post translational modifications,PTMs)的调节[6]。因此,对生物大分子结构与功能的深入研究不仅有助于揭示生命现象本质,更与人类健康息息相关,并将极大地促进药物靶点发现和新药研发。
在过去的40年中,结构生物学手段如X-射线晶体学(X-ray crystallography)[7]、核磁共振(Nuclear magnetic resonance spectroscopy,NMR)[8]、冷冻电镜(Electron cryomicroscopy,cryoEM)[9]、中子晶体衍射(Neutron diffraction)[10]和小角衍射(Small-angle scattering,SAS)[11]等技术的发展极大地推动了人们对蛋白质结构和功能的理解。不同的生物物理学方法各有优势和局限性。X-射线晶体学可以提供高分辨率(原子水平)的结构信息,但大分子量的蛋白复合物往往难以结晶。NMR能够在更接近于生理状态下获得蛋白质动态结构信息,但目前更多局限于分子量较小(<~50 kDa)的蛋白样品,且对蛋白质样品浓度要求较高。近年来,cryoEM技术的迅猛发展极大地拓展了人们对大蛋白复合物结构的认知,并且所需样品量少,可以获得较高分辨率的结构信息,但其分辨率往往受到样品异质性和分子量大小的影响。全面解析蛋白质或蛋白复合物结构和功能通常需要结合多种技术手段来实现,因此,互补技术的不断发展对于蛋白结构分析也起到了越来越重要的作用。
除了传统的生物物理学方法,质谱(Mass spectrometry,MS)技术也广泛应用于蛋白质领域的研究。1980年代末,软电离技术如基质辅助激光解吸电离(MALDI)和电喷雾电离(ESI)的发明推动了生物质谱的飞速发展,使得生物大分子能够以完整离子的形式被转移到气相中,从而被不同类型的质量分析器所检测[12]。质谱分析不仅可用于鉴定蛋白序列和翻译后修饰[13],还能用于研究蛋白质结构、折叠和动力学机制[14-16]。其中,氢氘交换质谱(Hydrogen deuterium exchange mass spectrometry,HDX-MS)技术是研究蛋白质结构动态变化的一种强有力工具,也是对传统生物物理手段的重要补充[17]。氢氘交换(HDX)用于研究蛋白质结构早已不是一个新的概念。Linderstrøm-Lang在1954年的工作中首次将蛋白质置于D2O中,利用密度梯度管测定氢氘交换速率从而研究蛋白质结构[18],并由此奠定了HDX的理论基础[19]。氢氘交换质谱技术可通过结合低温超高压高效液相色谱和质谱仪器分析确定酰胺氢氘交换的位置和速率。此外,实验分析流程和前期数据处理的自动化使得HDX-MS技术发展迅速,成为一种有效的蛋白结构分析工具[20]。虽然HDX-MS不能获得高分辨率的蛋白结构信息,但其优点在于基本不受分析体系的大小和复杂程度的限制[21],样品需求量少,并且还可用于研究对于传统结构学分析方法具有挑战的蛋白体系,如膜蛋白[22]、无序蛋白[23]和超大蛋白复合物[24]。本文首先介绍了HDX-MS的基本原理、交换机制、实验方法和技术研究进展,随后从蛋白质自身结构动态变化,蛋白质-小分子相互作用以及蛋白质-蛋白质相互作用3个方面对HDX-MS的应用进行介绍。

图1 骨架酰胺H(绿色)、侧链H(蓝色)和碳原子相连的H(黄色)
1 氢氘交换质谱技术概述
1.1 基本原理
HDX技术利用蛋白质中不稳定的氢原子可以与溶液中的氘原子发生交换的化学反应特性研究蛋白质结构[19],不同类型的氢原子H/D交换速率不同(图1)。例如,C—H键几乎不发生溶剂介导的交换;蛋白质N端和氨基酸侧链 (—OH,—SH,—NH)的H/D交换速率快,半衰期短(0.01~1 ms),并且交换上去的氘原子在分析后期很容易被溶液中的氢原子交换下来(回交,Back exchange),很难通过质谱方法检测[25];而骨架酰胺H/D交换速率相对缓慢,可以利用MS通过测定蛋白质的质量变化来监测交换反应。蛋白质的每个氨基酸(脯氨酸除外)都存在一个骨架酰胺氢,因此,骨架酰胺的H/D交换水平反映了蛋白质的构象变化。通过比较蛋白质在不同状态下氘代水平的变化,可以获得蛋白质结构和动态变化的信息。然而,主链酰胺氢交换速率受氢键、溶剂性质(pH值、温度)以及蛋白质构象动态变化的影响,可以发生显著变化[25-27]。
1.1.1 pH值氢氘交换是一类酸碱催化反应[28],酰胺氢的交换速率(kch)符合公式:
kch=kint,acid[H3O+]+kint,base[OH-]+kint,water[H2O]
(1)
其中,kint,acid和kint,base分别代表酸催化和碱催化的速率系数,kint,water则表示水催化质子转移的固有系数[28-29]。由公式(1)可知,酰胺氢交换速率高度依赖于环境pH值的变化。pH值低于2.5~3.0时,酸催化为主导;当pH值大于3.0时,质子转移主要受碱催化反应控制[30-31]。pH值位于2.5~3.0之间时,交换速率最小(约比pH 7.0时低105倍),称为pHmin。值得注意的是,生理环境下(pH 7.0~7.4),H/D交换几乎完全由碱催化控制,kint,acid和kint,water的影响可以忽略不计[29]。
1.1.2 温 度除pH值以外,温度也是影响交换速率的重要因素。生理条件下,HDX的活化能约为17 kcal/mol,环境温度每增加10 ℃,交换速率增加3倍[29,32]。根据公式(2)可以预测交换速率kch随温度变化的关系:
kch(T)=k293exp(-Ea/R[1/T-1/293])
(2)
其中,k293代表293 K时kint,acid、kint,base或kint,water的交换速率常数;Ea则是酸、碱或水催化HDX的活化能;R是气体常数0.001 986 kcal·mol-1·K-1。当温度从25 ℃降低至0 ℃,交换速率约降低14倍[29]。温度降低,分子动能和扩散速度减小,蛋白质骨架酰胺氢氘交换速率也随之减小。Quench条件下(pH 2.5,0 ℃),HDX速率最小,不易发生回交。因此,为了提高实验的重现性,实验过程中应精确控制pH值和温度,减少回交。
1.2 交换机制
上述因素均可以显著影响交换速率(高达两个数量级),但是对于折叠蛋白而言,蛋白质的高级结构对HDX速率影响更大,骨架酰胺氢交换速率主要受到氢键和溶剂可及性的影响[33]。若酰胺氢参与形成稳定的氢键或是位于蛋白质内部,则该位点酰胺氢交换速率非常缓慢。然而,当蛋白质构象发生变化时,酰胺氢的状态也会随之改变,交换速率因此而不同。
折叠蛋白的骨架酰胺氢原子与溶剂(D2O)交换遵循以下机制:
(3)
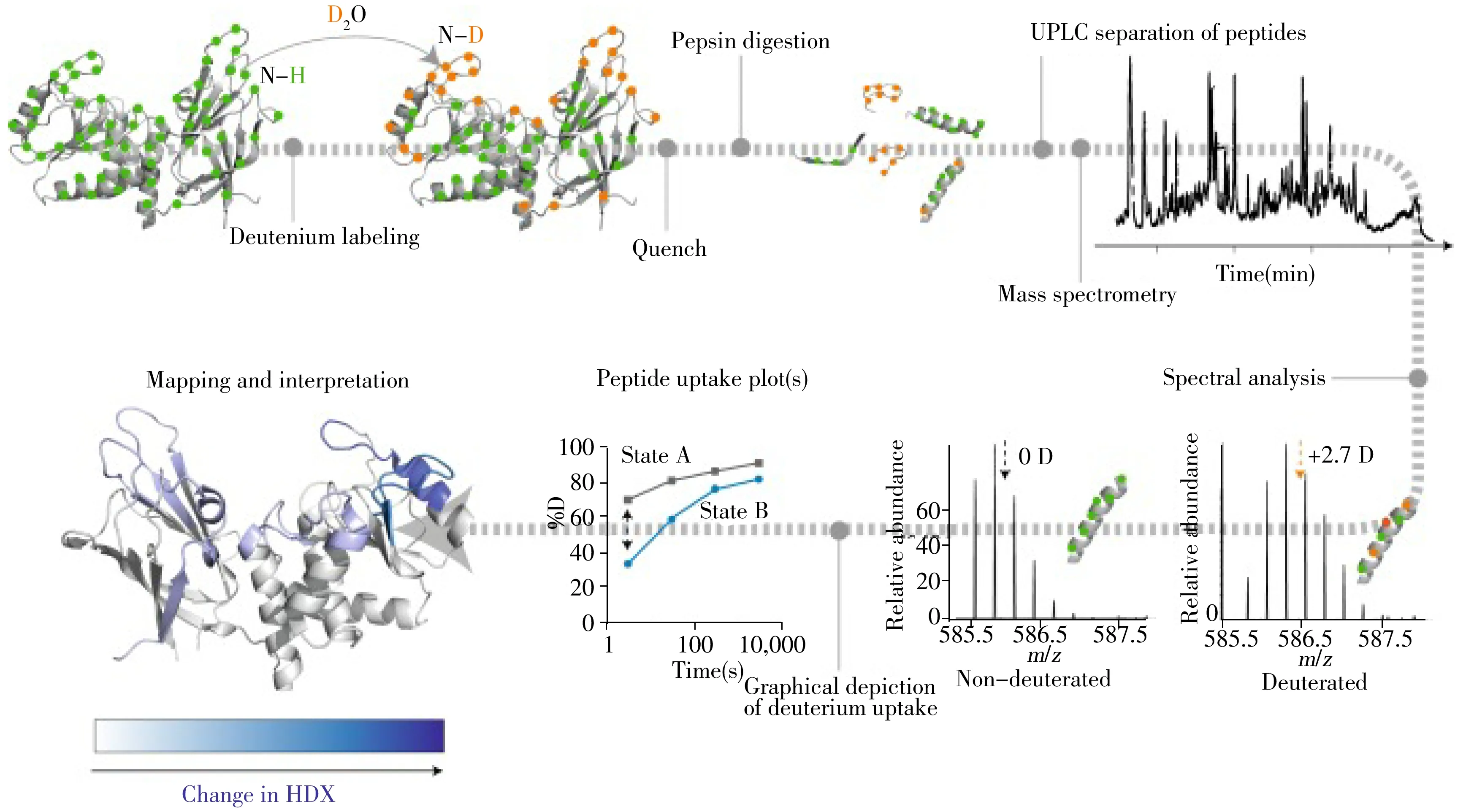
kop和kcl是蛋白质Opening和Closing状态的速率常数。当蛋白处于Opening状态时代表不参与形成氢键的酰胺氢,可与溶剂发生交换;相反,在蛋白处于Closing状态时,代表参与形成氢键的酰胺氢,位于蛋白质结构内部,不发生HDX。kch则是酰胺氢的固有交换速率常数。当kop>>kcl,交换速率主要受kch影响,此时蛋白质处于Opening状态。然而,当蛋白质处于一个类似于Native的构象状态时,kop< (4) 根据kch和kcl之间的相对速率,HDX分为EX1和EX2两种不同的交换机制[34-35]。当处于EX1动力学状态时(很少发生在Native构象),kcl< 根据实验体系和目的不同,HDX-MS实验可分为多个不同的实验步骤与方法,但任何HDX-MS实验方法都包含氘标记、氘代位点空间定位和数据处理与分析3大主要步骤。 氘标记可分为连续标记(Continuous labelling)和脉冲标记(Pulse labelling)两大类[36]。大多数HDX-MS实验采用连续标记策略。在连续标记方法中,蛋白样品置于重水(D2O)缓冲溶液中,但仍保持其天然构象(pD 7.4和生理缓冲条件)。蛋白质样品在氘代溶液中孵育一定时间(几秒到几小时不等),然后在pH 2.5和0 ℃条件下终止反应。这种策略能够监测氘代水平随时间的变化,提供蛋白质在平衡条件下的构象动态变化信息[37-38]。因此,连续标记常用于研究蛋白质与配体的结合、表位定位等。而在脉冲标记法中,蛋白质首先受到某种方式的干扰,如添加化学变性剂、改变pH值或温度,然后再将蛋白质置于氘代缓冲溶液中(pD值通常为8.0~10.0),在非常短的时间内标记(通常为10 s或更短)[37-38],因此HDX只发生在溶剂暴露的位置且不涉及氢键。脉冲标记可用于揭示蛋白质的折叠机制或去折叠时的构象变化过程。 氘标记之后,最直接的方法就是在完整蛋白水平测定HDX变化,通过将完整的蛋白质直接注入LC-MS系统中,测定蛋白质的总体质量数变化。蛋白质与不同的配体(蛋白、抑制剂等)孵育,H/D交换的差异得以可视化。但该策略不能提供任何氘代位点的信息及区域变化情况。目前常用bottom-up 策略实现氘代空间定位,蛋白质在氘水中孵育不同时间后,在pH 2.5和0 ℃的条件下终止反应,蛋白质经胃蛋白酶(pepsin)酶切为肽段进入LC-MS分析,氘的空间定位可以通过分析肽段的氘代水平来实现(图2)。实验流程主要分为以下5个步骤:(1)样品氘代:蛋白样品(蛋白复合物)在氘水(D2O)中孵育一定时间。(2)终止反应:pH 2.5,0 ℃。一般来说,实验中加入quench溶液调至pH 2.5,同时控制温度为0 ℃,可将HDX速率控制到最低。但根据不同的蛋白样品,quench溶液中也会加入不同浓度的变性剂(尿素或盐酸胍)和还原剂(三(2-羧乙基)膦,TCEP)帮助蛋白质变性还原,从而得到更好酶切效果,提高序列覆盖率[39]。(3)酶切:为了减少在后续的分析中发生回交,quench之后的所有步骤都应在pH 2.5和低温下尽快完成,再利用酸性蛋白酶将蛋白质酶切为肽段。实验中常用胃蛋白酶(pepsin),其他酸性蛋白酶如fungal,和nepenthesin也同样可用于HDX-MS[40-41]。(4)分离与质谱检测:氘代肽段通过低温HPLC/UPLC分离,可以在较短的时间内实现较好的分离并减少回交[42],然后通过电喷雾电离进入MS分析。根据氢氘之间的质量差异,MS可通过测定肽段质量中心位移来监测HDX变化。(5)数据处理:数据分析和结果阐述是HDX-MS分析的重要步骤。虽然目前的软件如Hexicon2[43]、HDWorkbench[44]、ExMS2[45]、HDExaminer(http://massspec.com/hdexaminer/)、HX-Express[46]、HDXFinder[47]、HDX-analyzer[48]、DECA[49]和Dynamx(https://www.waters.com/waters/library.htm?lid=134832928&locale=en_US)等可以帮助实现大部分的数据自动化分析,但是对所有谱图的手动检查仍然十分必要。 图2 Bottom-up HDX-MS实验流程图[50] 避免回交、提高实验准确度和重现性以及快速实现数据解析曾是HDX-MS技术广泛应用的瓶颈[51]。而实验流程和数据处理自动化的实现解决了这些难题,有效确立了HDX-MS技术在生物医药研发领域的应用地位。近年来,HDX-MS新技术和新方法的开发主要集中在拓展研究体系复杂性、提高结构分辨率和提升方法便捷性等方面。常规HDX-MS分析中,LC-MS峰容量的大小在一定程度上限制了所研究蛋白体系的大小和复杂程度。毛细管电泳技术(CE)不受传质阻力的限制和低温的不利影响,可以实现肽段的快速分离。Ramsey课题组发现,与LC进行的等效分离相比,CE分离复杂混合物的速度更快,峰容量几乎是前者的3倍[52]。Sheff等最新研发的低温纳流(500 nL/min)色谱系统仅需0.5 pmol的蛋白上样量就可以分析DNA-PKcs(469 kDa)的大型蛋白质复合物,极大地提高了系统的峰容量[53]。此外,离子淌度(Ion mobility spectrometry,IMS)技术的发展也为进一步提高峰容量提供了可能性。IMS可以在气相中实现多一维分离,从而可以大幅提高峰容量[54]。这些新技术的不断发展为拓展HDX-MS在复杂蛋白复合物体系中的应用奠定了良好基础。此外,目前利用HDX-MS获得的结构分辨率大多局限在肽段水平。串联酶柱的应用可以帮助获得更多更小的重叠肽,提升结构分辨率[55]。Hamuro等的研究也进一步证明采用串联酶柱可以达到单个氨基酸水平的结构分辨率[56]。同样,电子转移/捕获解离(ETD/ECD)串联技术因不会诱导H/D scrambling且断键均一的性质,为实现单个氨基酸水平分辨率提供了额外的选择[57-58]。串联酶柱和ETD/ECD方法高度兼容,二者联用是未来实现HDX-MS技术结构分辨率全面提升的不错选择。此外,受TECP难以快速实现二硫键完全还原的影响[59],富含二硫键的蛋白结构信息的获取往往有限。Mysling等发现电化学池可以在线快速还原蛋白二硫键,并可与HDX-MS兼容[60],该技术用于分析神经生长因子β时使序列覆盖率提高了近50%[61]。除传统的bottom-up策略,top-down还可以用于研究那些不适用于bottom-up分析的蛋白体系,如Aβ1-42淀粉样蛋白形成的构象动态变化[62]。最近,王冠博课题组将CE-HDX与top-down MS结合用于区分蛋白质的共存构象,并对构象的结构差异性进行表征[63]。 除本文介绍的液相HDX外,气相和固相HDX-MS近几年也逐渐兴起。气相HDX是一种快速(微秒反应时间)、灵敏的分析方法,可通过将蛋白侧链非骨架酰胺氢与高度碱性的气体ND3反应,探究蛋白侧链相互作用对蛋白质结构稳定性的影响[64]。Topp教授课题组开发的固相HDX-MS则可以用于蛋白质冻干制剂的表征[65]。在固相HDX-MS中,冻干粉中的蛋白质暴露在相对湿度和温度可控的D2O蒸汽中,氘代水平可以反映蛋白固相状态下,蛋白质-辅料相互作用以及蛋白质分子本身的构象变化[65- 66]。因此,HDX-MS技术的不断完善和发展,也使得HDX-MS对具有高度挑战性的复杂体系(如超大蛋白复合物[24,67-68]、高度异质性的糖蛋白[69-70]、膜蛋白[22]和无序蛋白[23]等)的分析成为可能。 理解蛋白质结构、动态变化和功能是结构生物学的核心,也是深入了解体内生理和病理过程的先决条件。因此,围绕蛋白的结构、构象动态变化以及靶向药物筛选、设计和作用机制等进行研究,对于阐明蛋白在生理和病理过程的作用和推动新药研发至关重要[71]。HDX-MS能够在蛋白质或蛋白复合物溶液体系下观察蛋白构象动态变化、配体结合并确定配体结合区域,而且还能观察伴随结合信息的蛋白质构象动力学差异,其快速、灵敏的检测特性可作为潜在的高通量药物筛选平台。 蛋白结构的动态变化往往与其功能息息相关,表征这种结构动态变化对于理解结构与功能之间的关系至关重要。其中,生理环境的变化可以改变蛋白结构和构象,从而影响蛋白的生理学功能,如流感病毒膜表面的血凝素(HA)蛋白在酸性条件下发生结构变化释放融合肽,使得病毒与细胞发生膜融合从而感染宿主。X-射线晶体学虽然提供了HA不同状态下的晶体结构,但是难以捕捉HA在体内酸性条件下的膜融合结构变化过程。Garcia等采用HDX-MS首次研究并证明酸性条件下HA融合肽的释放应该在HA1松动和HA2弹簧加载重新折叠之前[72]。这一研究为阐明膜融合病毒的机制以及药物设计提供了结构基础。同样,蛋白合成过程的不同结构状态也影响其生理学功能,例如,神经生长因子β(NGF)对神经系统的正确发展至关重要。NGF具有两种形式:成熟的NGF和proNGF。这两种形式对神经元有相反的作用:NGF诱导增殖,而proNGF则通过与常见的神经营养素受体(p75NTR)结合诱导凋亡。尽管NGF的结构已得到解析,但是proNGF的结构细节一直未知。Trabjerg等的研究结果表明,proNGF的前体肽段存在一个局部的高级结构。通过对比二者HDX-MS交换数据发现,proNGF前体肽段的存在对其结构中的3个loop区域起到稳定作用。此外,该课题组在proNGF的前体肽段中还发现了两个N糖和两个O糖修饰[73]。这些结果提高了对proNGF和NGF构象特性的认识,为在分子水平上阐明两者的不同生物学效应提供了结构基础。 除具有确定结构的蛋白质外,无序蛋白(Intrinsically disordered proteins,IDPs)也是重要的潜在药物设计靶点,在细胞信号传导、调控和癌症中发挥关键作用[74]。对比研究发现,IDPs比同等长度的有序蛋白具有更多的结合腔,为靶向药物设计提供了可能性[75]。然而,受其结构特殊性的困扰,往往难以用X射线方法得到IDPs的晶体结构。尽管NMR是研究蛋白动态结构的利器,但无序区域的NMR共振信号通常高度重叠难于解析。近年来,HDX-MS在IDPs结构研究中的应用逐渐兴起,并提供了重要结构信息。例如,表皮生长因子受体(EGFR)在乳腺癌等癌症中起重要作用,是重要的药物靶点。Keppel等采用HDX-MS研究了酪氨酸激酶EGFR、HER2和HER3 3种受体,发现与有序的激酶结构域相比,C末端尾部吸收氘的速度更快,从而证明这3种蛋白质的C末端结构域在很大程度上是无序结构[76](图3)。而Iacob等的研究结果进一步表明这些受体C末端结构域的无序和扩展构象有助于增加信号伴侣的捕获半径[77]。这一发现对这些受体酪氨酸激酶(RTKs)的下游信号蛋白和信号传导的理解具有重要意义。 除此之外,HDX-MS还广泛应用于生物制药生产和开发过程中的高阶结构表征和质量控制。蛋白质药物的生产依赖于细胞表达,生物合成过程中产生的不同程度的PTMs是诱导药物三维结构和功能发生变化的重要因素[78-79]。Houde等采用整体和局部HDX-MS技术对PTMs(如甲硫氨酸氧化、盐藻糖糖基化和半乳糖糖基化等)如何影响重组单克隆抗体IgG1的构象动态变化进行了研究。以糖基化结果为例,整体HDX分析显示IgG1具有高度稳定的高级结构,而局部HDX研究则显示糖基化区域去糖基后氘代水平发生改变,说明糖基化会影响抗体对受体的识别[80-81]。同样,HDX-MS还可用于表征复杂且异质性高的生物药物体系,如重组因子IX-Fc融合蛋白[82]。 图3 EGFR、HER2和HER3的HDX-MS曲线[76] 图4 氘代30 s(A)、300 s(B)和15 h(C)后,卡拉洛尔与β2 AR结合的HDX结果映射到β2 AR T4L晶体结构(PDB:2RH1)[84] 生理条件下,蛋白质结合小分子配体发挥生物学功能。因此,研究蛋白与小分子化合物的作用机制,识别小分子结合和变构区域,观察靶蛋白结构动态变化,有助于阐明蛋白结构和功能之间的动态关联及药物作用机制。G蛋白偶联受体(G protein-coupled receptors,GPCRs)是目前研究最为广泛的药物靶点之一[83]。GPCRs是最大的细胞膜表面信号蛋白家族,可将细胞外信号传递到细胞内以维持细胞内稳态。这种传导通过配体诱导的构象变化来实现,并通常伴有受体远端区域的结构变化。Griffin课题组以β2-肾上腺素能受体(β2-AR)膜蛋白为例,通过优化HDX-MS的实验参数和选择适宜的去污剂,有效提高了蛋白序列覆盖率。该研究分析了在拮抗剂卡拉洛尔作用下β2-AR的配体依赖性构象变化(图4),提供了受体的高度动态区域和化学修饰信息[84]。后续研究分别在不同配体(从完全激动剂到拮抗剂)存在的情况下,阐述了β2-AR的构象动态变化[85]。此工作为后续研究膜蛋白提供了方法指导,并证明了HDX-MS用于测定蛋白功能的实用性。 与GPCR一样,离子通道也是药物作用的重要靶点之一。所有的钠离子转运体(NSSs)都存在结合与不结合配体两种状态之间的构象动态平衡,但现有研究结构都不能很好地解释转运体这两种状态之间的结构转变及运输机制。Rand课题组最近先后研究了亮氨酸转运体(Leucine transporter,LeuT)、5-羟色胺转运体(Human serotonin transporter,SERT)和多巴胺转运体(Dopamine transporter,DAT)3种转运体两种状态之间的构象动态变化,为深入理解NSSs功能背后的分子机制奠定了基础[86-88]。 HDX-MS还可以通过研究小分子与靶蛋白的作用机制进一步辅助药物研发与设计。例如,腺嘌呤核苷酸(AMP)依赖的蛋白激酶(Adenosine 5'-monophosphate (AMP)-activated protein kinase,AMPK)是生物能量代谢调节的关键分子,也是研究代谢疾病的重要靶点。Griffin课题组利用HDX-MS发现AMP与AMPK结合主要引起γ亚基构象变化,α和β亚基变化微小。小分子激动剂A769662则结合在α和β亚基之间,两种配体激活AMPK的分子机制不同[89]。这些数据为设计和发现用于治疗AMPK相关代谢疾病提供了研究依据。 此外,在基于片段药物设计寻找先导化合物的方法中,HDX-MS也可以作为潜在的高通量筛选平台。基于配体调节维生素D受体(VDR)在骨质疏松症的口服治疗中具有潜在应用价值,Carson等利用HDX-MS识别片段与VDR的结合区域,对潜在的结构片段进行筛选,发现一些同天然激素VD3的结合区域类似的片段,这些信息可以辅助先导化合物的发现及其结构优化[90]。Anand课题组采用HDX-MS对Hsp 90 N端ATPase结构域与高亲和力配体(KD≈20 nmol/L)和低亲和力片段(KD≈500 μmol/L)结合的互作进行了研究,发现了配体与片段的结合区域和远端变构区域[91]。此外,与传统药物筛选平台相比,研究还发现两个结构母核相同、亲和度(Kd)相近的低亲和力片段之间的氘代水平也存在显著差异,由此证明HDX-MS可以用于低亲和力化合物的结构筛选。 蛋白质-蛋白质相互作用(Protein-protein interactions,PPIs)参与了众多细胞内生理过程,如信号传导、分子转运和各种代谢途径等。而异常的PPIs是导致多种疾病发生的原因,识别体内的PPIs作用界面有助于药物设计与开发。IL-23R及其同源细胞因子IL-23形成的复合物与自身免疫性疾病相关。Sayago等利用HDX-MS确定了IL-23R和IL-23结合区域,并发现二者结合后,IL-23R的N端免疫球蛋白结构域出现明显的氘代水平降低现象。基于针对PPIs互作界面设计药物的思路,他们进行了多肽抑制剂的设计,并结合计算机模拟技术确定了多肽与IL-23R的结合作用位点,实现了靶向PPI界面设计药物的目的[92]。 生物体内的蛋白往往以多蛋白复合物形式发挥生理功能。因此,针对多蛋白复杂体系的结构研究有助于对蛋白功能的进一步理解。GroEL/ES伴侣蛋白系统可以促进和增强底物蛋白的折叠途径,但机制尚不清楚。Georgescauld等采用HDX-MS分析DapA(GroEL依赖的TIM桶状蛋白)自发折叠和辅助折叠过程,发现DapA自发折叠需要较长的时间,而通过GroEL/ES催化可将折叠加速30倍以上[68]。由此说明GroEL依赖的TIM桶状蛋白需要通过GroEL/ES进行折叠催化才能在生物学相关的时间尺度上达到天然状态,从而避免聚集或降解。 此外,HDX-MS还广泛应用于抗体-抗原互作表位的识别,这对抗体药物作用机制的阐明尤为重要。白喉类毒素(DT)抗原是儿科和增强联合疫苗的主要成分之一,然而,对白喉毒素(DTx)表位的结构解析以及潜在的抗体中和机制尚无报道。Zhu等利用HDX-MS和生物膜层干涉技术(BLI)识别两种单克隆抗体(mAbs)在白喉毒素上的特异性表位,结果表明两种mAbs的作用机制完全不同。单克隆抗体2-25选择性地与DTx的b亚基结合,相反,2-18与a亚基结合[93]。这一结果阐明了抗体中和机制,并有助于揭示影响白喉疫苗疗效的潜在因素。HDX-MS还可以用于研究复杂的抗原表位定位[94-100]。例如,HIV包膜糖蛋白(Env)是中和抗体的唯一靶点,但目前已报道的结构大多是广泛中和抗体(bnab)与截断的Env亚基或组分形成的复合物。它们与完整的Env三聚体之间的相互作用以及在生理环境中形成中和抗性结构的决定因素尚未得到确定。Guttman等采用HDX-MS首次研究并报道了一组广泛中和抗体(Broadly neutralizing antibodies,bNAbs)和native-like Env三聚体之间的相互作用(SOSIP.664三聚体)。如图5所示,VRC01 (IC50=0.32 μg·mL-1)和b12(IC50=2.82 μg·mL-1)都是识别CD4结合位点(CD4 binding site,CD4bs)的抗体,高特异性VRC01 Fab只在其结合界面产生局部效应,而低特异性b12 Fab结合则与整个三聚体的复杂变化有关(结合界面显示为绿色)。结果表明,弱效的抗体只能在三聚体短暂的开放状态下结合[100]。不仅如此,HDX-MS技术还可以研究单个抗体与蛋白复合物的结合机制或是多个抗体与单个抗原的作用机制。VHH6是一种识别IL-6-gp80复合物但不单独识别单个蛋白亚基的抗体,Adams等利用HDX-MS技术发现VHH6的CDR区域与IL-6和gp80同时相互作用,将两者锁定在一起,这一结果为筛选和结构辅助药物发现提供了新的视角[101]。Zhang等则采用HDX研究4个非竞争性抗体单独或联合使用对天然花粉过敏原蛋白(Bet v1)的作用机制。HDX结果表明,4种抗Bet v1抗体保护了Bet v1的特定区域,从而解释了单克隆抗体与多克隆抗体IgEs结合Bet v1阻断效率的差异。作者首次应用HDX阐明了联合抗体与单独抗体治疗Bet v1诱导过敏的优势,为后续药物设计提供了新思路[102]。 图5 靶向CD4bs的bNAb结合的影响[100] 尽管HDX-MS技术在国外药物研发流程中广泛应用,但受技术细节、数据解析、重现性等多因素的限制,目前HDX-MS在国内生物医药研究领域的应用还较少。因此,建立规范化的、具有行业共识的实验流程和方案,进一步提高数据自动解析度,建立蛋白质动态信息中央HDX数据库是全面推广HDX-MS技术的关键。此外,串联酶柱、液相-气相分离技术的结合以及与ETD/ECD解离多种技术的串联应用也将是未来提升HDX-MS结构信息分辨率的发展趋势。而在此基础上,采用针对性开发的统计学方法从HDX数据中提取出保护因子(Protection factor)则有望全面实现HDX-MS技术的单氨基酸分辨率[103]。而气相或是在电喷雾界面发生的HDX-MS将拓展我们对蛋白侧链氢如何参与氢键和盐桥作用,稳定蛋白结构的认识。最后,HDX-MS与其他生物质谱技术如交联质谱(Cross-linking mass spectrometry,CX-MS)[104]、非变性质谱(Native mass spectrometry)[105]和共价标记质谱(Covalent labeling mass spectrometry,CL-MS)[106]联用,也同样将拓展和加深我们对蛋白质/蛋白复合物结构和动态信息的认识,为靶向药物设计提供结构和方法学基础。1.3 HDX-MS实验方法
1.4 HDX-MS技术研究最新进展
2 HDX-MS在蛋白及蛋白复合物研究中的应用进展
2.1 蛋白自身结构动态变化
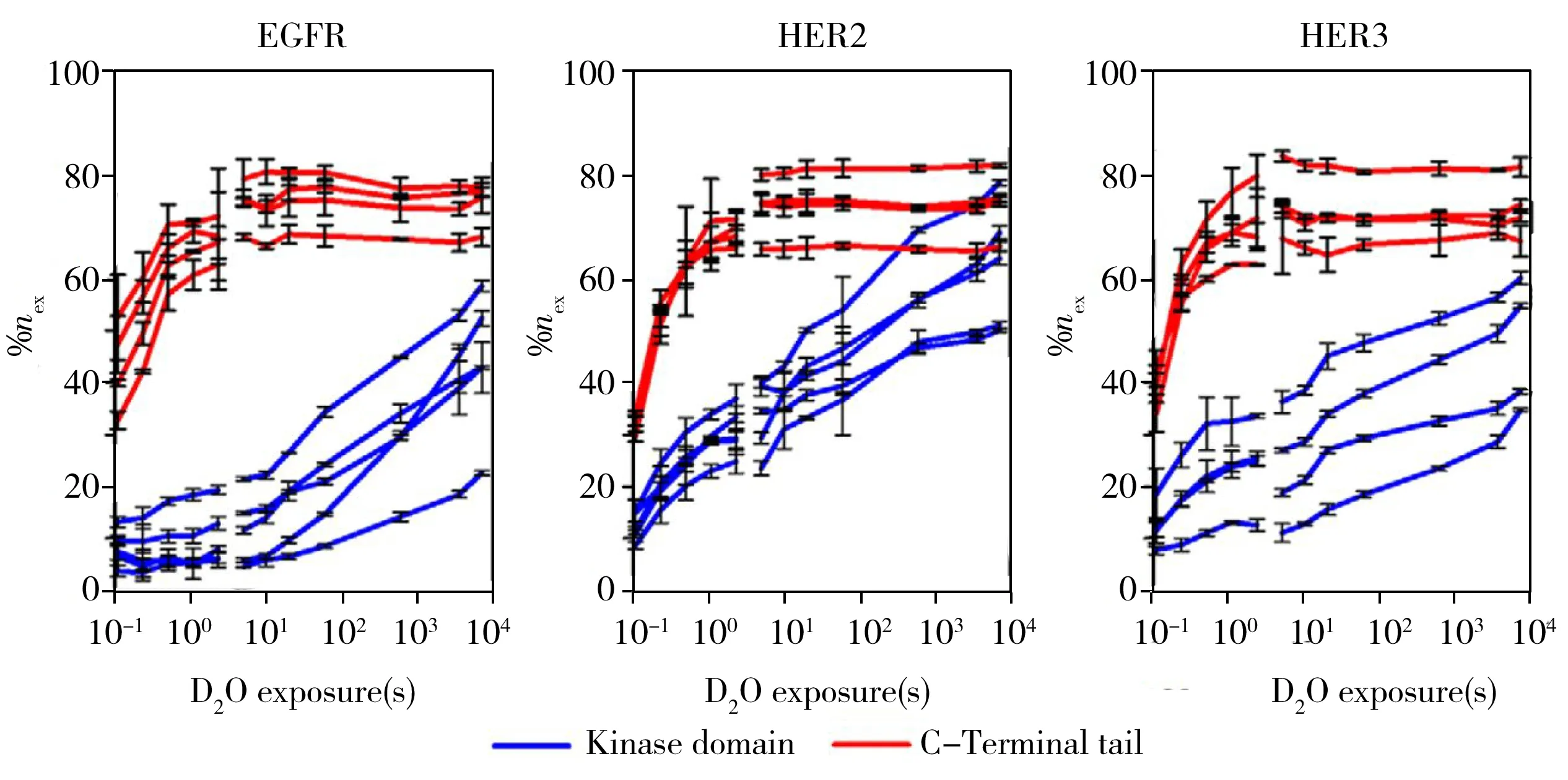
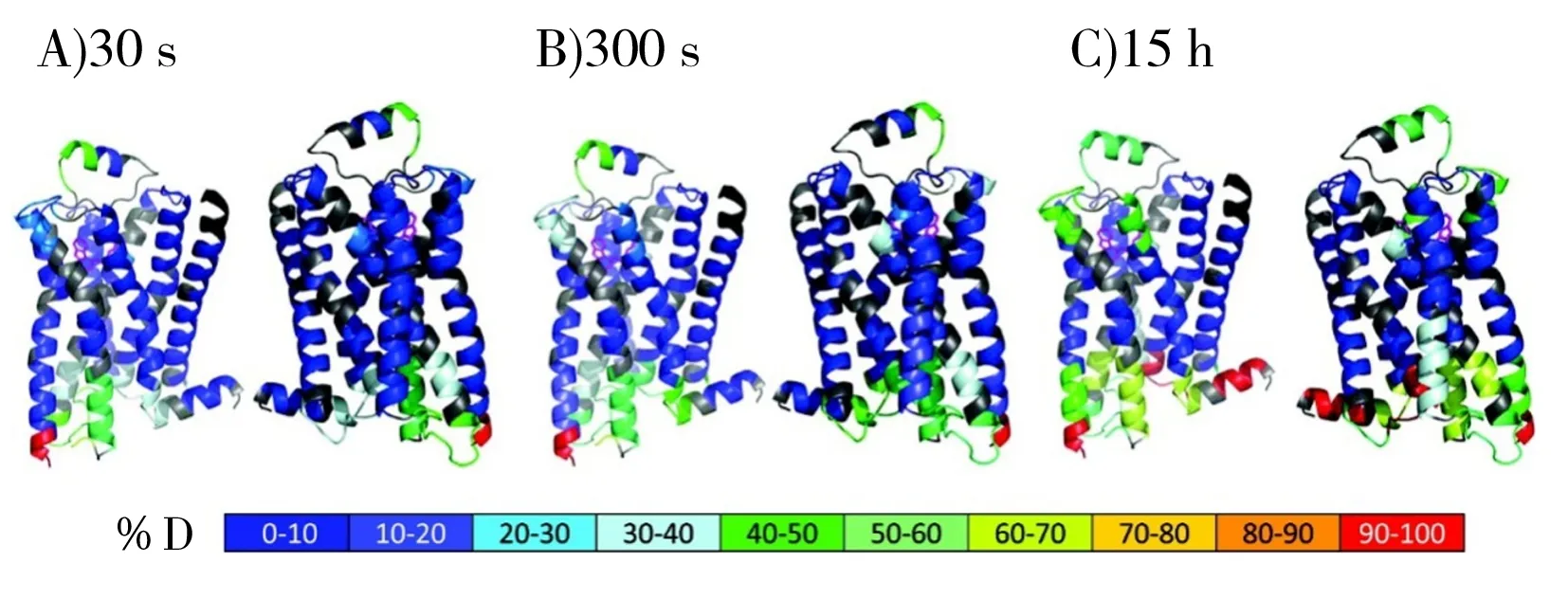
2.2 蛋白质-小分子相互作用
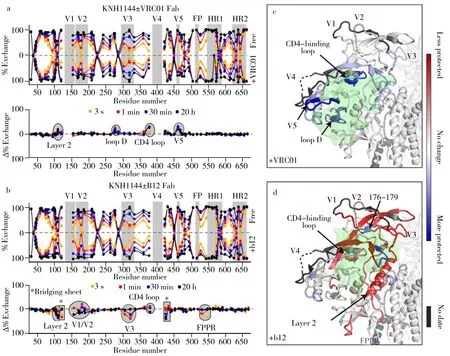
2.3 蛋白质-蛋白质相互作用
3 总结与展望
