基于液相色谱-质谱联用技术的代谢组学分析方法研究进展
2020-02-28徐天润刘心昱许国旺
徐天润,刘心昱,许国旺
(1.中国科学院分离分析化学重点实验室 中国科学院大连化学物理研究所,辽宁 大连 116023;2.中国科学院大学,北京 100049)
代谢组学是研究生命体受到病理生理刺激或基因环境扰动后,糖类、脂质、核苷酸和氨基酸等内源性小分子代谢物(分子量小于1 000)种类和数量的变化[1-2]。相比于其他组学,代谢组学反映生命体已经发生的生物学事件,因此能够更准确直接地反映生命体终端和表型信息[1]。目前,广泛应用于代谢组学数据采集的技术平台有氢/碳核磁共振技术(1H,13C-NMR)、气相色谱-质谱技术(GC-MS)、液相色谱-质谱技术(LC-MS)、毛细管电泳-质谱技术(CE-MS)以及直接进样质谱技术(DIMS)等。鉴于代谢物种类多样且浓度差异大,代谢组学研究需要依托高灵敏度、高分辨率的分析技术。色谱-质谱联用技术具有灵敏度高、选择性好、通量高等优势[3],能够根据代谢物检测的需求选择不同的电离模式和质量分析器。丰富的多级质谱数据有助于代谢物的结构鉴定,从而最大限度地获取代谢物信息。图1统计了2015年~2019年8月期间基于色谱-质谱联用的代谢组学论文发表数量,可以看出液相色谱-质谱联用技术(LC-MS)已成为代谢组学最常用的技术平台[4],论文数量稳步增长。
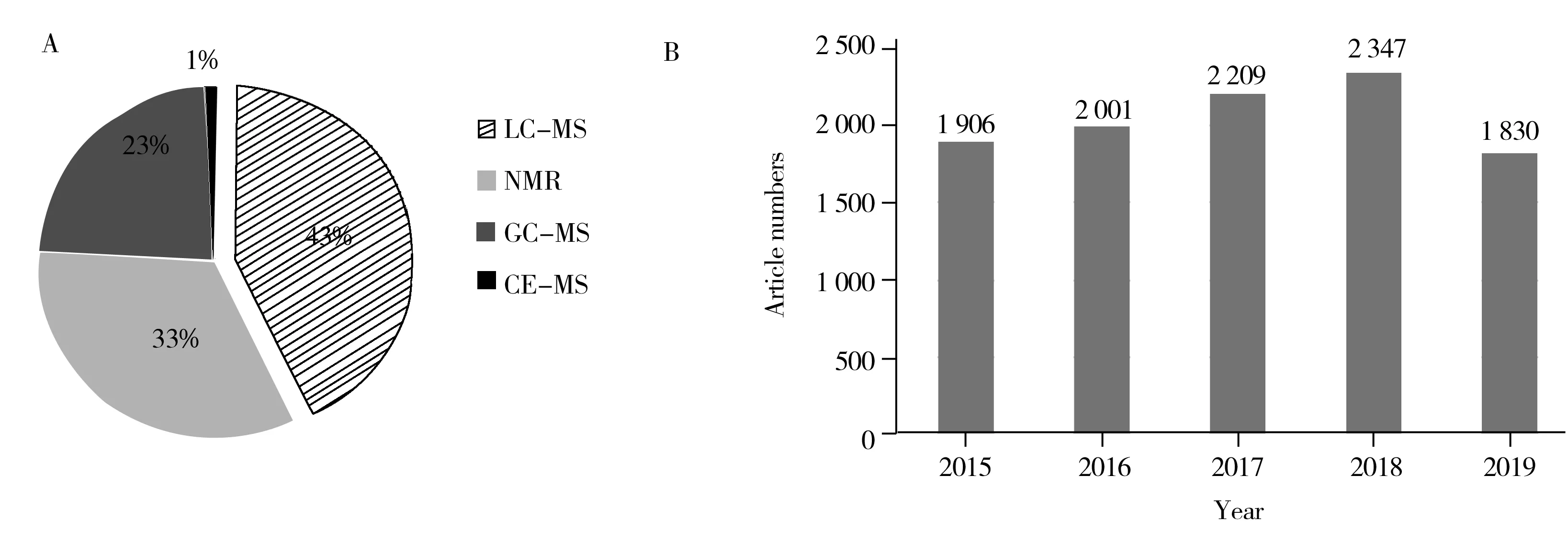
图1 基于GC-MS、LC-MS、CE-MS、NMR的代谢组学论文数量对比(A),以及基于LC-MS的代谢组学论文数目对比(B)
代谢组学研究的流程一般包括样品采集、样品前处理、代谢物的分离分析、数据处理(包括数据预处理、数据分析及代谢物鉴定)和代谢物的生物学功能阐释。目前,人类代谢组数据库(Human metabolome database,HMDB)已给出超过11万种代谢物,但对于生物体内代谢物的总数仍不清楚。代谢物全面的定性、定量分析面临较大挑战,在代谢组学方法上需要不断创新。在预处理方法上,要尽可能充分地提取和富集低丰度代谢物;在检测上,要不断扩大代谢物的检测覆盖度,并提高分析通量;在深入挖掘代谢组学数据的同时还需注重代谢组学与其他组学数据的整合,进而深化对相关代谢途径的解释和认知。唯有确保每一个环节均能创新发展,才能最终推动代谢组学技术取得巨大进步。本文将重点阐述近5年来基于LC-MS的代谢组学分析方法的研究进展。
1 代谢组学液相色谱分离方法的进展
1.1 一维色谱
随着高效固定相的研制和装柱技术的改进,液相色谱(LC)的柱效不断提高。高压液流系统的应用,使得细粒径固定相的使用得以普及,大大提高了液相色谱的分离能力,也加快了分析速度。色谱固定相的选择应与被分离物的物化性质相匹配,以实现高选择性和良好分离度。目前应用最多的液相色谱是反相液相色谱(RPLC),主要用于分离非极性或中等极性代谢物。Panopoulos等[5]利用反相液相色谱-四极杆-飞行时间质谱(RPLC-Q-TOF-MS)分析胚胎干细胞中代谢物的相对丰度,检测到超过5 000个代谢物特征,展现了反相液相色谱-质谱联用技术强大的分离与检测能力。为提高分离效率,可在流动相中添加改性剂以改善分离度[6]。Bertram等[7]评估了常用添加剂(如0.1%甲酸、乙酸铵、氟化铵等)对LC分离效果的影响,结果表明0.1%甲酸并不能提供最佳的分离效果。1 mmol/L乙酸是负离子模式下尿样代谢组学研究的最佳添加剂,相比于0.1%甲酸,检测到的峰数量和信号强度均有显著提升。因此正、负电离模式下使用单一的添加剂往往不能获得最佳的代谢物覆盖,通常需要两种不同的添加剂。
与RPLC互补,亲水相互作用色谱(HILIC)已成为强极性和亲水性代谢物的替代选择[8]。大量研究人员对不同HILIC固定相的柱效和保留行为进行了评估,结果表明两性离子柱ZIC-HILIC可提供最佳性能[9-11]。Zhong等[12]在正、负离子模式下采用HILIC和RPLC柱分析唾液的代谢特征,以筛查乳腺癌的潜在生物标志物。研究发现,HILIC可有效分离在RPLC上死时间出峰的强极性代谢物,基于两个色谱柱组合的数据提供了更好的分型和预测结果。为改善HILIC柱保留时间在代谢物峰匹配中的可靠性,Feng等[13]提出了计算HILIC保留指数的方法,用于校准保留时间的漂移,以提高代谢物注释的准确性。
在色谱填料进步的同时,超高效液相色谱(UPLC)的问世大大拓宽了液相色谱的应用领域。UPLC可耐受超高压力(15 000~18 000 psi),结合较小的粒径(亚2 μm颗粒)及超高柱效填料(窄内径色谱柱),可实现样品的高效、快速分离并获得尖锐的色谱峰。与传统液相色谱相比,UPLC具有缩短分析时间和提高分离度两大优势。前者从节约成本的角度,可在短时间内分析大批量样品,后者对复杂样品的分析极为重要。Gray等[14]将色谱柱内径由2.1 mm缩小至1.0 mm,使所有分析物的响应提升了2~3倍,溶剂用量减少75%。由于信噪比的提升,该方法能够实现分离性能和稳健性的显著增益。UPLC与高分辨质谱联用可使组学分析数据在分辨率、灵敏度和通量方面更强大。将超高效液相色谱-四极杆-飞行时间质谱(UPLC-Q-TOF-MS)用于分析茶叶的代谢组,峰对齐后检测到674个特征离子[15]。该平台也应用于抑郁症患者的血浆代谢组学分析,正、负离子模式下分别获得4 724和4 504个特征离子[16]。
1.2 多维色谱
多维液相色谱(Multi-dimensional liquid chromatography,MDLC)的概念最早由Giddings 等[17-18]提出,是指样品中的组分经过两次或多次LC分离,且被分开的组分在后续分离中不会再融合。与传统LC方法相比,MDLC技术可结合不同的分离机理增加分离维数和提高峰容量,在提高覆盖度和分离度方面具有独特优势。
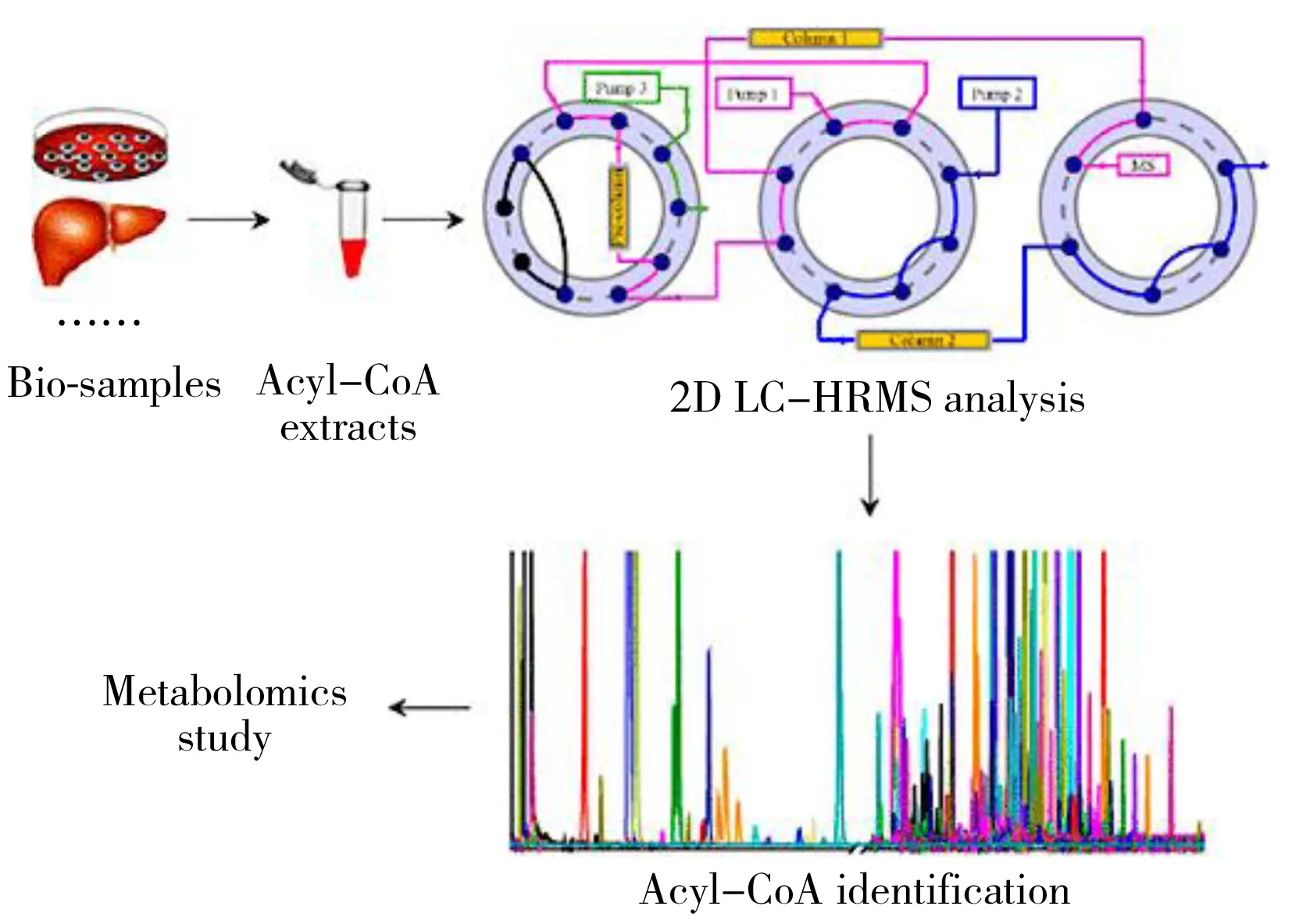
图2 在线二维液相色谱-质谱法用于短链、中链和长链的酰基辅酶A的分析[29]
本课题组在二维及多维色谱方面开展了系统的研究。考虑到对弱极性或中等极性化合物全二维液相色谱的分离方法相对成熟[19],Wang等[20]建立了针对极性化合物分离的HILIC×HILIC全二维系统。Yang等[21]建立了在线的Ag+LC×RPLC 全二维流路用于分析甘油三酯,第一维和第二维分别根据不饱和度与碳链长度实现分离。相比于一维方法,选择性得到提高,但峰容量未显著提升,原因在于两维流速差距较大,存在稀释效应[22]。为解决这一问题,Wang等[23]发展了在线停留HILIC-RPLC系统,一方面用稀释液降低第一维出口溶剂的强度,另一方面引入捕集柱将第一维流出的馏分较好地保留在前端,通过第二维流动相反吹将馏分转移至第二维,有效地降低了稀释效应,实现了较低的进样宽度。应用于血浆的脂质组学分析中,第一维实现了脂质族分离,第二维根据脂肪链进行精细分离,该模式的峰容量高达415,与全二维分析取得的实际峰容量相接近。在线停留模式还应用于3D LC的构建,第一维的预分离将复杂样品分为几个不同馏分,经停留接口对各馏分进行有序的全二维分析,有效改善了复杂样品的分离效果[24]。针对传统分离方法覆盖范围不广的问题,Wang等[25]建立了基于柱切换的分析系统,通过将极性不同的HILIC和RPLC色谱柱结合,将HILIC柱上非保留的疏水性代谢物洗脱并转移至RPLC柱上分离,扩展了代谢物的覆盖范围,使单柱模式下死时间流出的代谢物得以保留,实现了极性和非极性代谢物的依次分析,也可以通过配置两个检测器实现同时分析[26]。应用于大鼠尿液的代谢分析中,共检测到5 686个极性代谢物离子和1 808个非极性代谢物离子,体现了方法在复杂样品分离分析中的适用性[27]。为进一步提高代谢物覆盖度,通过预柱分离,将代谢物在线切割为中等极性和弱极性两个馏分,Wang等[28]分别进行代谢组学和脂质组学分析,对血浆的全组分分析共鉴定出447种(ESI+)和289种(ESI-)代谢物和脂质,使一次分析同时获得代谢组和脂质组信息成为可能。将该流程稍加改进应用于酰基辅酶A(Acyl-CoA)的分析(图2),实现了一次进样同时涵盖短链、中链和长链的酰基辅酶A的分析[29]。
多维色谱的应用越来越广泛,很多课题组开展了很好的工作。Liu等[30]建立的NP/RP 2D LC-MS方法实现了脂质类物质的同时检测,应用于良性、恶性乳腺肿瘤患者血浆的全脂质分析,成功鉴定了512种脂质。该课题组还将方法用于腔隙性脑梗[31]、结直肠癌[32]和动脉粥样硬化[33]脂质组学分析,证实了NP/RP 2D LC-MS实现脂质类物质全覆盖的巨大潜力。HILIC和RPLC分离系统提供了两种完全不同的保留机制和非常高的正交性,可以实现一次进样同时分离检测复杂样品中的亲水和疏水性化合物,提高峰容量和分离通量[8]。Ivanisevic等[34]将RPLC-ESI+和HILIC-ESI-模式结合构建正交的二维体系,该方法可全面涵盖中心碳代谢通路中的代谢物(如氨基酸、有机酸、磷酸化糖等)信息。Contrepois等[9]的研究表明将RPLC-MS和HILIC-MS方法结合,可使尿液和血浆代谢组扩增44%,比单独使用RPLC-MS增加了108%的代谢物特征,大大提高了代谢组覆盖率。
2 基于LC-MS的代谢组学分析方法进展
LC-MS具有高灵敏和高度稳健的双重优势,从而可提供良好的代谢物覆盖率[35-37],是代谢组学研究中应用最广泛的技术平台,比GC-MS更具灵活性和普适性[38-40]。在实际样品分析中,LC-MS采用的策略包括非靶向、靶向和拟靶向3种。
2.1 基于LC-MS的非靶向代谢组学
非靶向分析是最经典、应用最广泛的代谢组学方法。通过对样本中所有化合物的综合、全面、无偏性的高覆盖检测,可实现不同样品间代谢特征的比较。因此,样品制备应尽可能保持代谢组提取的完整性,所采用的方法应具有非选择性、快速和可重复性。分辨率对于非靶向分析至关重要,结合高扫描速度和高质量精确度的要求,高分辨质谱Q-TOF、Q-Orbitrap(四极杆-轨道离子阱)是提供这些性能的最佳选择[41],获得的高分辨质谱信息有助于快速检索以鉴定代谢物。Raro等[42]评估了Q-TOF和Q-Orbitrap对非靶向代谢组学确定兴奋剂睾酮滥用标志物的性能,两个质谱仪虽然检测到的特征数量不同,但均显示相同的潜在标志物。因此这两种平台对于非靶向代谢组学的研究同样有效,均适用于未知化合物鉴定。非靶向分析通常在正离子和负离子模式下分析相同样品,可提供更广泛的代谢物覆盖[43]。Breitkopf等[44]利用具有正/负离子模式切换的高分辨质谱鉴定出来自18种脂质类别和66个亚类的共2 493个脂质,其中正离子模式下鉴定1 756个,负离子模式下鉴定737个,单次LC-MS/MS即可获得两种电离模式的质谱信息。
传统的非靶向方法分析1个样品通常需要半小时,这大大限制了分析通量。因此,快速、高通量的非靶向方法尤为重要。应用短柱或微柱、提高色谱柱温度及流速是缩短洗脱梯度、进而缩短分析时间的良好选择。Gray等[14]将传统的2.1 mm×100 mm色谱柱替换为1 mm×50 mm色谱柱,直径和长度的缩小使谱带展宽最小化,性能提升的同时减少了溶剂用量。将该方法应用于小鼠的尿液样品分析,结果表明尽管峰容量与检测到的特征数量比传统方法少,但仍可筛查不同样本之间关键的差异代谢物[45]。为兼顾分析速度与峰容量,Ouyang等[46]建立了在12 min内完成代谢谱采集的快速方法,节省了约60%的分析时间,适合于大规模样本的非靶向代谢组学研究。短梯度的UPLC-MS方法有利于提高分析通量,节省分析时间,但由于色谱分离度降低导致代谢物覆盖率的损失是高通量非靶向方法的弊端,这也不利于发现具有生物学功能的痕量代谢物,因此,还需要根据分析需求选择不同的分析方法。
2.2 基于LC-MS的靶向代谢组学
靶向分析是对特定代谢途径相关的少数已知靶向分子进行精准定量。靶向分析依赖于串联质谱固有的高选择性和高灵敏度,使用三重四极杆(QQQ)质谱仪在多反应监测(MRM)模式下检测代谢物的前体离子和特征产物离子,通常被认为是“金标准”[47-48]。Han等[49]建立了在负离子模式下的人血清中50种胆汁酸的MRM数据库。Yan等[50]建立了直接耦合的RPLC-HILIC方法进行靶向分析,鉴定出人体尿液中198个代谢物。使用三重四极杆-线性离子阱(Q-Trap)构建MRM策略的研究亦有报道。Chen等[51]在Q-Trap平台上获得了698个水稻叶片代谢物的二级质谱信息,定性了135种代谢物,相对定量了277种代谢物。
由于提供MRM模式的QQQ、Q-Trap的分辨率较低,限制了其在复杂系统生物学研究中的应用,因此需要开发新的方法以拓宽靶向代谢组学的研究途径。基于高分辨、高精度Q-Orbitrap的平行反应监测(PRM)模式的靶向定量策略应运而生,相比于MRM,PRM能实现高分辨地全面检测目标代谢物的所有碎片离子。依托PRM扫描模式,Zhou等[52]开发了一种针对237种代谢物的大规模定量方法,并与全扫描和MRM的性能进行了系统比较。由于碎裂过程赋予PRM更高的选择性,相比于全扫描其具有更高的准确度,相比QQQ具有更高的分辨率。对复杂生物样本的评估显示出PRM对靶向代谢物定量的优异性能,可以作为MRM靶向分析的补充策略[53]。PRM还可以辅助定性,许国旺课题组[54]开发了一种新策略,大大提高了酰基肉碱的检测覆盖率。通过全扫描MS/ddMS2模式和PRM模式的结合层层递进采集MS数据,用以识别特征碎片信息。进一步整合子类同源分组和预测结果,共得到758种酰基肉碱的信息,这是目前最全面的酰基肉碱列表。全面的靶向分析得益于两步数据采集提供的充足的MS结构信息,使得全面鉴定生物样本中的酰基肉碱成为可能。
传统的靶向分析代谢物覆盖度及通量有限,无法满足对时间成本和经济效益的需求,因此需要发展快速、高通量的靶向方法。QQQ仪器实现了高扫描速度和快速正负离子模式切换的功能,并且单次转换停留时间不超过5 ms,使得MRM分析成为快速、高通量靶向分析的理想策略[53]。Yuan等[55]采用HILIC正负离子切换模式,实现了单次进样15 min即可得到289个MRM离子对,定性分析了258个代谢物。Wei等[56]将3种色谱条件和两种电离方式配置在一个体系中,利用柱切换的时间间隔进行柱清洗和平衡,实现了仅用10 min靶向定量包括氨基酸、核酸和有机酸在内的205种内源性代谢物,以快速的方式实现了最佳分离。鉴于高分辨质谱的低扫描速度,PRM在分析通量方面很难与MRM匹敌。因此,MRM和PRM两种模式各有利弊,但结合具有完整MS2采集的PRM和具有快扫速及电离模式切换的MRM,将为快速、高通量靶向代谢组学定量提供强大的工具。
综上,由于重复性和稳定性有限,非靶向分析难以为大规模样品分析提供高质量数据。靶向分析由于使用有限的代谢物标准品,限制了代谢物覆盖度的提升。两种方法各有优劣,往往需在代谢物覆盖度和数据质量间进行权衡,导致会损失很多重要的代谢物信息。因此,需要开发代谢组学新技术以提供高覆盖度、高质量的数据。
2.3 基于LC-MS的拟靶向代谢组学
结合非靶向方法的高信息覆盖度及靶向方法的高灵敏度、高重复性的优势,许国旺课题组首次提出了拟靶向代谢组学的概念[57-58]。该方法首先通过高分辨质谱全面获取代谢物的一级和二级质谱信息,从二级质谱中挑选特征离子对,随后在具有MRM模式的QQQ仪器上对离子对进行靶向采集。相比于靶向分析,拟靶向方法通过采集混合生物样品(QC)的二级质谱代谢特征,可以同时包括定性与未定性的代谢物,实现了广泛覆盖。与非靶向分析相比,MRM模式的数据稳定性高、线性范围宽[58],能够获得高质量的数据信息,可以关注到更多低丰度代谢物[59]。
拟靶向方法的关键是获取特征离子对,二级质谱信息的全面获取是MRM离子对覆盖度的重要影响因素。基于信息依赖采集(IDA)模式采集二级质谱信息,应用本课题组自主开发的自动化获取特征离子对软件MRM-Ion Pair Finder,共检测到854个代谢物离子对[60],具有很好的代谢物覆盖率和可靠的生物标志物鉴定潜力。顺序窗口采集所有理论碎片离子(SWATH)模式的二级质谱信息采集能力优于IDA,在正离子模式下,从血浆标准参考物质(NIST SRM 1950)中获得1 373个具有离子对的代谢物,与IDA相比,SWATH可以采集更多的离子对[61]。除此之外,对于有碎裂规律的化合物(如脂质),可以根据保留时间和碎片与脂质结构的规则进行预测。Xuan等[62]建立了高覆盖度的拟靶向脂质组学分析方法,共涵盖19个脂类、3 377个脂质离子对,覆盖7 000多种脂质分子结构,与靶向脂质组学方法相比,显示出更高的脂质覆盖度,适合于大规模脂质组学分析(图3)。在上述基础上,本课题组也建立了基于LC-MS拟靶向方法的多批次数据校正方法,保证了大规模、大批量代谢组学数据的稳健可靠[63-64]。

图3 基于UPLC-MS的拟靶向脂质组学[62]
除了拟靶向分析方法,“广泛靶向”和“全局优化靶向(GOT)”等新型代谢组学技术也得到发展,为传统的代谢组学分析提供了有价值的补充方法。Chen等[51]基于Q-Trap质谱仪发展了“广泛靶向”代谢组学技术,通过自建的MS数据库,采用MRM模式扫描,匹配物质参数信息进行定性和定量。代谢物覆盖的广度取决于物种特异的MS库覆盖具有生物学功能代谢物的全面性,只有获得生物样本最全面的代谢物信息,才能具有广泛的代谢物覆盖能力。Gu等[65]建立了对血清代谢物具有广泛覆盖的GOT-MS方法,为了提高亲水性代谢物的覆盖度,采用选择离子监测(SIM)增强扫描(60~600 Da),选择具有良好峰形且信噪比S/N> 3的离子作为母离子,在MRM模式下进行MS/MS碎片扫描,最终获得针对595个母离子的1 890个MRM离子对。GOT-MS的关键是利用QQQ对母离子和子离子进行全局扫描,优化和扩展了QQQ的检测能力,实现了广泛的代谢物覆盖。以“拟靶向”、“广泛靶向”和“全局优化靶向”为代表的新一代代谢组学技术,还将更深层次地结合非靶向的“广泛性”和靶向的“精确性”,以期获得更全面、更精准的生物样品代谢谱信息。
3 基于LC-MS的化合物定性方法
在LC-MS代谢组学研究中如何大规模准确定性代谢物仍然是个极具挑战性的难题。广泛采用的策略是将代谢物的质谱数据与数据库比对,通过精确分子量(一级质谱)和/或二级质谱信息实现定性。该方法存在的主要问题如下:(1)由于代谢物众多,即使质谱的质量精度小于1 ppm[66],仍不足以准确推断元素组成;(2)数据库中来自于待测代谢物的二级质谱图太少,影响了检索的命中率。加之LC-MS流动相、基质、离子源等对化合物碎裂的影响,使得基于数据库检索更是难上加难。到目前为止,对大多数生物样本,只能对4%~5%的LC-MS峰进行定性[67]。
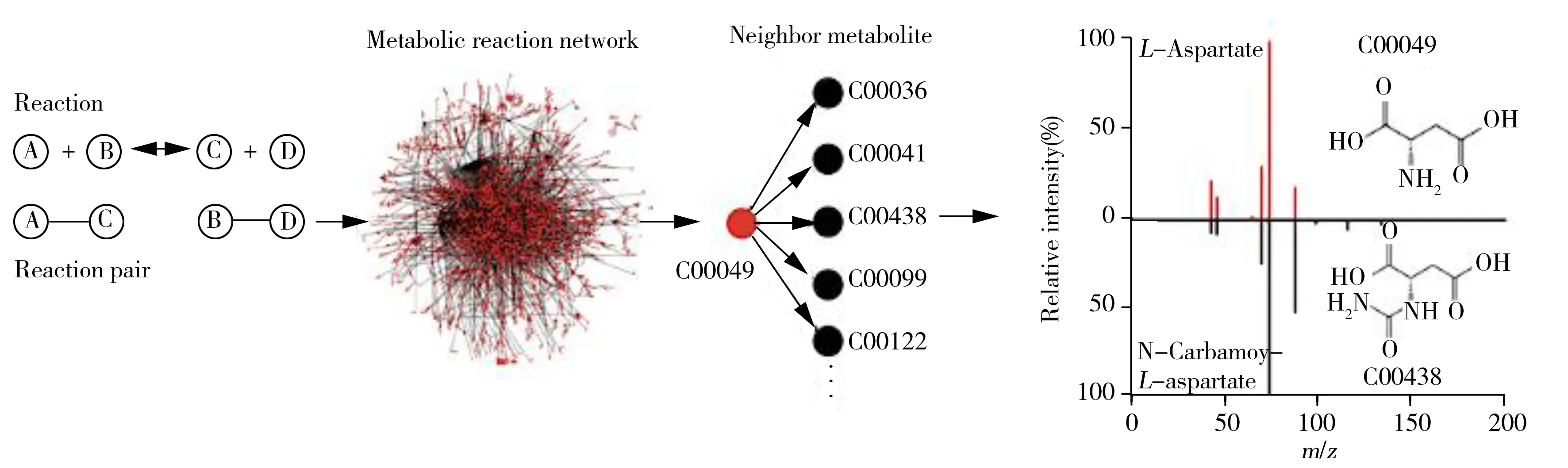
图4 代谢反应网络的构建策略[71]
尽管如此,近年来,关于代谢物定性的工作仍取得了一些进展,主要集中在以下3个方面。一是基于碎片质谱数据推测代谢物结构。对于有碎裂规律的分子类别(如脂质),通过收集不同类别脂质的碎裂和重排信息建立数据库,根据碎裂规则可预测分子结构[68]。为提高预测精度,可以应用机器学习从实验数据中训练模型参数。Allen等[69]提出了竞争性碎片模型(CFM),该模型根据数据学习碎裂过程,通过预测任何碎裂过程的可能性,给出最有可能观察到的峰,从而提高预测的精确度。机器学习还可用于预测化合物的分子指纹。Shen等[70]将碎片树和机器学习相结合预测分子指纹并用于分子结构鉴定,显著提高了代谢物鉴定的能力。二是基于反应物和产物在结构上存在关联,利用二级谱与结构相似性的内在联系鉴定未知代谢物。预测模型MetDNA[71]选择种子代谢物绘制代谢反应网络,并利用代谢物间二级质谱的点积来评估代谢物在反应网络中是否相邻,逐步扩展注释代谢物,实现了单次实验约2 000个代谢物的注释,置信水平高达90%(图4)。预测模型iMET[72]使用各种碰撞能量下代谢物间的二级质谱的余弦相似性和精准质量差异来预测代谢物在反应网络中的上下游关系。为提高注释方法的自动化能力,采用机器学习从原始数据集中训练模型,实现自动对预测结构打分排序,未知化合物鉴定的有效性取决于现有数据库的覆盖范围,因此融合更多数据库对训练更准确的模型至关重要[73]。通过二级质谱的相似性来注释结构相关的代谢物,大大增强了现有数据库的鉴定能力。三是基于稳定同位素标记推断化学结构,其中最为常用的是13C、15N、34S[74]。通常情况下,将标记的生物样品与天然类似物混合,通过有效计数代谢物中标记的原子数,显著减少可能的分子式数量,进而改善代谢组学数据的生物学解释[75]。对方法的改进是采用独特的同位素峰分布,如5%或95%的13C标记,即同位素比值异常分析(IROA)技术[76-77],可使识别和量化代谢物的能力明显增强。
4 多组学整合技术平台
对代谢途径的深入阐释仅依靠代谢组学数据还不够,应有多组学数据。多组学数据可以更全面地理解生物学机制。
多组学整合通过多层次分析代谢通路来探寻生物调控的分子机制,Lee等[78]对细胞样品进行了蛋白质层面和代谢层面的分析,通过分析代谢物和代谢酶的表达水平的相关性,揭示了细胞增殖过程中代谢通路的调控机制。Wettersten等[79]采用蛋白组学-代谢组学相结合的分析方法研究不同分级的胃癌组织,揭示了多条调控胃癌分级的信号通路,有助于不同分级胃癌的个性化治疗。多组学数据的整合分析会使研究更加系统,分析结果既能互为印证,又能互为补充。Geiger等[80]从蛋白组和代谢组两个水平进行了大规模非靶向筛选,分析T细胞激活与非激活状态下蛋白组和代谢组的差异,发现L-精氨酸代谢通路中的蛋白质和代谢物均发生显著变化,进一步的细胞功能实验验证了L-精氨酸对T细胞代谢网络的调控机制。Gupta等[81]研究了植物激素对大豆叶片蛋白质和代谢物的影响,发现与类黄酮和异类黄酮生物合成的蛋白丰度增加,代谢组的结果与蛋白组一致。
多组学技术平台的整合依赖于分析方法、系统生物学和计算机技术的共同进步。在分析方法上,需要建立更加先进的分析手段以高通量地获取稳定可靠的数据,为系统生物学提供科学基础;在计算机技术上,应深入挖掘数据并提供高效的数据处理和计算方法,实现对相关代谢途径的系统认知。
5 结论与展望
随着人们越来越认识到代谢研究的重要性,代谢组学得到了愈加广泛的应用,包括疾病诊断、潜在生物标志物鉴定、疾病发病机制阐明和代谢新通路揭示等,尤其是基于LC-MS的代谢组学技术更是突飞猛进。在色谱分离方面,UPLC能够实现在短时间内对复杂样本的更高效分离;2D LC结合化学计量学的巨大进步能够大大提高代谢特征检测量,实现代谢物的全面覆盖。在质谱分析方面,对于非靶向分析,高分辨质谱已必不可少,尤其是与UPLC联用;基于MRM模式的三重四极杆质谱被认为是靶向分析的“金标准”,基于PRM的高分辨质谱亦逐步应用于靶向代谢组学分析,并显现出优异的定量能力;结合非靶向和靶向分析优势的拟靶向技术显示出与非靶向相接近的代谢物覆盖度及与靶向相当的数据质量,得到了日益广泛的应用。
基于LC-MS的代谢组学尚存在以下待解决的问题:(1)大样本的代谢组学研究需要更加快速、稳健、重复性更高的分析方法,从而以更高的准确度检测代谢特征的差异;(2)对样本量极少的代谢组学研究,需要发展超高灵敏的痕量代谢物检测方法以实现代谢物的广泛覆盖;(3)仍需加强对代谢物的大规模准确定性以推动代谢物的生理学解释;(4)亟待开发生物信息学工具以加速多组学数据整合。虽然目前的LC-MS技术远达不到代谢组学的需求,但相信随着科研人员孜孜不倦的钻研和代谢组学的深入发展,其必将会在疾病诊疗中发挥更大作用。
