Rapid identification of chemical profile in Gandou decoction by UPLCQ-TOF-MSE coupled with novel informatics UNIFI platform
2020-02-28LiXuYiLiuHongfeiWuHunWuXiochungLiuAnZhou
Li Xu ,Yi Liu ,c,Hongfei Wu ,Hun Wu ,Xiochung Liu ,An Zhou ,,*
a The Experimental Research Center,Anhui University of Chinese Medicine,Hefei 230038,China
b Anhui Province Key Laboratory of Chinese Medicinal Formula,Hefei 230012,China
c Waters Corporation(China),Shanghai 201206,China
Keywords:
ABSTRACT
1.Introduction
Traditional Chinese medicines(TCM)have been extensively used for the prevention and treatment of complex and chronic diseases in China[1,2].TCM formulae,combination of medicinal plants or animal materials,collectively exert therapeutic actions by complex interactions among multiple components from different herbal medicines.Based on TCM theories,these constituents in formulae could play a multi-target,synergistic and harmless therapeutic role[3].As the components in TCM are rather complicated,it is difficult to separate and identify multiple chemical constituents.Therefore,developing a rapid and reliable method for elucidating the composition of TCM is necessary.
Wilson's disease(WD),also known as hepatolenticular degeneration,is an autosomal recessive genetic disorder of copper metabolism caused by ATP7B gene mutation[4,5].Excessive copper accumulation in patients suffering from WD leads to liver disease,neurological disorder,K-F rings,and osteoporosis[6].Currently,there are several chelating agents such as D-penicillamine,dimercaptosuccinic acid,trientine,and tetrathiomolybdate for medical therapy[7].Although Western conventional medications are highly effective,prevalent,and low-priced,a number of side effects have been observed with chelation therapy[8].Gandou decoction(GDD),a classical TCM formula,has been used in clinics to treat WD for decades in China[9,10].It is composed of six crude drugs,i.e.,Rheum palmatum L.(Da-Huang),Coptis chinensis Franch.(Huang-Lian),Curcuma longa L.(Jiang-Huang),Lysimachia christinae Hance(Jin-Qian-Cao),Alisma orientale(Sam.)Juzep.(Ze-Xie)and Panax notoginseng(Burk.)F.H.Chen(San-Qi).The clinical studies have been proven that GDD can promote urinary copper excretion,ameliorate liver function and improve the patient's clinical symptoms[7,11].Furthermore,GDD appears to be safe,effective,and well tolerated and has fewer adverse effects than Western conventional medications[12].In our previous studies,we investigated the therapeutic effect and serum metabolic profiling of GDD in copper-laden rats.It was found that GDD could reduce the hepatic copper accumulation,and improve liver pathological characteristics by restoring the impaired lipid metabolism,amino metabolism and glucose metabolism[13].However,due to multi-component systems of TCM,the chemical constituents of GDD still remain unclear.Therefore,a systematic chemical profiling research of GDD is in an urgent need.
In recent years,UPLC-Q-TOF-MSE(where E represents collision energy)has provided a powerful approach for the efficient separation and structural characterization of TCM with the advantage of its high resolution,sensitivity and accuracy[14].Q-TOF-MSEcapable of simultaneously acquiring accurate mass precursor ion in MS full scan and fragment ions in MSEhigh-energy scan increased the credibility of analysis results[15,16].Additionally,UNIFI software from Waters Corporation is a versatile and automated data processing platform.The software incorporates scientific library into a streamlined workflow to integrate data acquisition,library searching,MS fragment matching and report generation,which alleviates the workload from massive MS data and realizes rapid analysis of chemical components[17].This high throughput strategy was innovatively used for screening and identification of chemical components in herbal medicines[18,19]and TCM formulae[20].In the present study,an integrative strategy based on UPLC-Q-TOF-MSEcoupled with UNIFI informatics platform has been applied to reveal the chemical profile of GDD.The aim of this study is to develop an analytical method for elucidating the material basis of GDD and provide valuable information for the quality control and in vivo analysis.
2.Material and methods
2.1.Materials and reagents
Rheum palmatum L.,Coptis chinensis Franch.,Curcuma longa L.,Lysimachia christinae Hance,Alisma orientale(Sam.)Juzep.and Panax notoginseng(Burk.)F.H.Chen were purchased from Beijing Tongrentang Co.,Ltd.(Hefei,China)and authenticated by Doctor Rongchun Han(College of Pharmacy,Anhui University of Chinese Medicine,Hefei,China).All voucher specimens were deposited at the authors'laboratory.The reference standards,including berberine hydrochloride,physcion,emodin,alisol B 23-acetate,quercetin and notoginsenoside R1,were obtained from the National Institutes for Food and Drug Control(Beijing,China).Chrysophanol,rhein,aloe-emodin and kaempferide were obtained from Beina Chuanglian Biotechnology Research Institute(Beijing,China).Curcumin was isolated in our laboratory with a purity of more than 98%by HPLC,and its structure and molecular weigh have been identified by using several spectral analyses and MS,respectively.Acetonitrile and methanol(LC-MS grade)were purchased from TEDIA(Fairfield,USA).Formic acid was obtained from Tianjin Guangfu Fine Chemical Research Institute(Tianjin,China).Ultrapure water was purified using a Milli-Q water purification system(Millipore,Billerica,MA,USA).
2.2.Standards and sample preparation
GDD consisted of six ingredients,including Rheum palmatum L.(20.0 g),Coptis chinensis Franch(20.0 g),Curcuma longa L.(20.0 g),Lysimachia christinae Hance(24.0 g),Alisma orientale(Sam.)Juzep.(24.0 g)and Panax notoginseng(Burk.)F.H.Chen(3.0 g).They were mixed together and immersed in 0.8 L distilled water(1:8,w/v)for 0.5 h.Afterwards,they were decocted twice by extracting and refluxing for 1 h each time.Finally,the two extractions were combined and concentrated to 1.0 g crude drug per milliliter,and then the solution was freeze-dried and stored in a vacuum desiccator before use.The accurately weighed 1.0 g freeze-dried powder was dispersed in 30 mL methanol and ultrasonicated in a water bath for 30 min to prepare solutions.The individual preparation of six herbs was carried out according to the same procedures as that of GDD.An aliquot of 5μL filtrate was injected into the UPLC-Q-TOFMSEsystem for analysis after filtered through 0.22μm filter membrane.
11 reference standards were dissolved in methanol.Before qualitative analysis,they were mixed together to make reasonable concentration and filtered through 0.22μm filter membrane.
2.3.Chromatography and mass spectrometry conditions
Chromatographic analysis was performed using a Waters Acquity™UPLC system(Waters Corporation,Milford,USA).Chromatographic separation was carried out at 30℃,using an Agilent Eclipse Plus C18 RRHD column(2.1 mm×100 mm,1.8μm)with mobile phases A(0.1%formic acid in water)and B(acetonitrile).The flow rate was set at 0.3 mL/min.The gradient profile was as follows:0-1 min,10%-10%B;1-4 min,10%-20%B;4-10 min,20%-30%B;10-15 min,30%-40%B;15-18 min,40%-50%B;18-23 min,50%-75%B;23-25 min,75%-85% B;25-27 min,85%-100% B.
Mass spectrometric detection was carried out on Waters Xevo G2 Q-TOF mass spectrometer(Waters Corporation,Milford,USA)equipped with an ESI source.The full scan data were acquired from 50 to 1200 Da,using a capillary voltage of 3.0 kV for positive ion mode and-2.5 kV for negative ion mode,sampling cone voltage of 40 V for positive ion mode and 50 V for negative ion mode,extraction cone voltage of 4.0 V,source temperature of 120℃(ESI+)or 110℃(ESI-),cone gas flow of 50 L/h,desolvation gas(N2)flow of 600 L/h and desolvation gas temperature of 350 ℃.The collision voltage was set as 6.0 eV for low-energy scan and 20-80 eV for high-energy scan.Data were centroided and mass was corrected during acquisition using an external reference(Lock-Spray™)consisting of a 200 pg/mL solution of leucineenk ephalin infused at a flow rate of 10μL/min via a lockspray interface,generating a realtime reference ion of[M+H]+(m/z 556.2771)in positive ion mode and[M-H]-(m/z 554.2615)in negative ion mode to ensure accurate MS analysis.All data collected in centroid mode were obtained and used to calculate the accurate mass and composition of relative target ions with MassLynx™V4.1 software(Waters).
2.4.Establishment of a chemical compounds library of GDD
The systematic information on chemical compounds isolated from the six individual herbs in GDD was collected and sorted out by retrieving databases such as China Journals of Full-text database(CNKI),Medline,PubMed,Web of Science and ChemSpider.A selfbuilding library of chemical compounds was established by UNIFI software,including compound name,molecular formula,chemical structure,and accurate molecular mass.Among them,the information of 356 compounds is listed in Table S1.
2.5.Data analysis by UNIFI platform
All MS data analysis was processed on the platform of UNIFI software.Minimum peak area of 200 was set for 2D peak detection.The peaks intensity of high energy over 80 counts and the peak intensity of low energy over 200 counts were the selected parameters in 3D peak detection.A margin of error up to 5 ppm for identified compounds was allowed and the matching compounds would be generated predicted fragments from structure.We selected positive adducts including H+,Na+and negative adducts containing HCOO-and H-.They were allowed cross adduct combinations.
3.Results and discussion
3.1.Identification and characterization of chemical compounds
The high resolution MS data of GDD were quickly acquired by UPLC-Q-TOF-MSEmethod.The base peak intensity(BPI)chromatograms of GDD in positive and negative ion modes are depicted in Fig.1.The UNIFI screening platform was utilized to process and analyze the MS data,and then automatically matched the fragment information.After further manual verification,a total of 96 compounds were identified or tentatively characterized in GDD,including 21 anthraquinones,14 alkaloids,17 protostane triterpenoids,10 flavonoids,8 triterpenoid saponins,10 tannins,4 curcuminoids and 12 others.The detailed MS information of these components is summarized in Table 1.Meanwhile,the chemical structures were confirmed based on accurate mass,MSEdata and related literatures.The structures of main chemical constituents in GDD are shown in Fig.2.
3.2.Analysis of GDD by UPLC-Q-TOF-MSE
3.2.1.Anthraquinones

Fig.1.The base peak intensity(BPI)chromatograms of GDD from UPLC-Q-TOF-MSE analysis.(A)Negative scan;(B)Positive scan.
21 anthraquinones were detected from GDD and were also the major bioactive constituents of Rheum palmatum L.In this study,6 free anthraquinones,13 anthraquinone glycosides and 2 anthrones were determined based on MS database-matching.Anthraquinones have a characteristic fragmentation behavior with successive or simultaneous losses of CO,OH,CH3and CO2[21,22].Peaks 64,65,76,83 and 86 were exactly identified as aloe emodin,rhein,emodin,chrysophanol and physcion by comparing retention time and fragmentation patterns with reference standards.Rhein,the main anthraquinone in GDD,was used to characterize the fragmentation pathways(Fig.3).Rhein showed quasi-molecular ion[M-H]-at m/z 283.0256 in negative ion mode,and yielded fragment ions at m/z 239.0356 and 211.0403 by losses of CO2and CO,respectively.And then,the ion at m/z 211.0403 could further lose one molecule of CO to generate ion at m/z 183.0439.Aloe emodin and emodin were isomers with the same[M-H]-ion at m/z 269.In high energy MSEspectra,emodin revealed[M-H-CO]-ion at m/z 241.0507 and[M-H-CO-O]-ion at m/z 225.0559,while aloe emodin could be differentiated by the characteristic ion[M-HCHO]-at m/z 240.0421.Physcion showed[M-H]-ion at m/z 283.0619,and the obvious fragments ions at m/z 255.0315[M-HCO]-and 240.0359[M-H-CO-CH3]-were further obtained.Chrysophanol showed[M-H]-ion at m/z 253.0515,only one product ion at m/z 225.0545[M-H-CO]-.
For anthraquinone glycosides,aglycone ions were identified based on the MS fragmentation behaviors of free anthraquinones.Peak 47 exhibited[M-H]-ion at m/z 415.1045,which generated an[M-H-162Da]-ion at m/z 253.0515 by eliminating the glucose residue.The further loss of CO was in accordance with the characteristic ion at m/z 225.0563 of chrysophanol.Thus,peak 47 was assigned as chrysophanol-1-O-β-D-glc.Based on these fragmentation patterns,peaks 15,17,32,35,44,45,46,47,48,51,52,53,and 60 were inferred as anthraquinones glycosides.In addition,anthrones are an important type of anthraquinone.Sennosides usually gave a significant ion at m/z 386 which originated from C-10-C-10′cleavage.Peak 27 showed[M-H]-ion at m/z 861.1908,which first produced ions at m/z 699.1358 and 655.1880 by sequential loss of terminal glucose residue and CO2,followed by the cleavage of C-10 and C-10′forming[M-H-Glc-CO2-C15H9O5]-ion at m/z 386.1002.Its structure was identified as sennoside A or B.These two isomers were not distinguished from each other only by their MS spectra.Similarly,peak 25 displayed the same fragmentation patterns as peak 27,so it was presumed as sennoside C or D.
3.2.2.Alkaloids
A total of 14 alkaloids were identified in positive ion mode and came from Coptis chinensis Franch,including protoberberine alkaloids,apomorphine alkaloids,and tetrahydroprotoberberine alkaloids.As reported in the literature,the neutral losses like the methyl radical(CH3·),hydrogen radical(H·)and CO are the main fragment patterns of protoberberine alkaloids due to the successive cleavage of substituted methoxyl or methylenedioxyl groups on the A-and D-rings[23].Peak 39 was unequivocally identified as berberine by contrast with a reference standard.The MS spectrum and possible fragmentation pathways of berberine are depicted in Fig.4.Taking berberine as an example,it produced fragment ions at m/z 321.0983[M-CH3]+,320.0918[M-CH3-H]+,306.0763[M-2CH3]+,292.0969[M-CH3-H-CO]+and 278.0813[M-2CH3-CO]+.Peak 40 showed[M]+ion at m/z 352.1538 and yielded characteristic ions at m/z 337.1430[M-CH3]+,336.0983[M-CH3-H]+,322.1068[M-2CH3]+and 308.1275[M-CH3-H-CO]+,which indicated that it was presumed as palmatine.Likewise,the other peaks 31,33,34,38 and 67 could be tentatively identified as coptisine,jatrorrhizine,epiberberine,worenine and 8-oxoberberine,respectively.
Peak 10 gave[M]+ion at m/z 342.1701 with a molecular formula C20H24NO4.The predominant ions appeared at m/z 297.1123[M-(CH3)2NH]+,282.0876[M-(CH3)2NH-CH3]+,265.0861[M-(CH3)2NH-CH3OH]+,and 237.0908[M-(CH3)2NH-CH3OH-CO]+,which is consistent with the common structure of apomorphine alkaloids.Thus,peak 10 was considered as magnoflorine.Analogously,peaks 8 and 11 were deemed as isocorydine and isoboldine.Additionally,the tetrahydroprotoberberine alkaloids have retro-Diels-Alder(RDA)reaction,resulting in the cleavage of theterminal chain,such as-CH3.Peak 26 gave a protonated ion at m/z 356.1865.The fragment ions at m/z 192.1019 and 165.0893 were attributed to RDA cleavage at C-and B-rings.By further loss of a methyl radical,two obtained ions generated characteristic ions at m/z 177.0774 and 150.0663,respectively.Therefore,peak 26 was tentatively identified as tetrahydropalmatine.

Table 1 Identifciation of chemical constituents of GDD by UPLC-Q-TOF-MSE.

Table 1(continued)
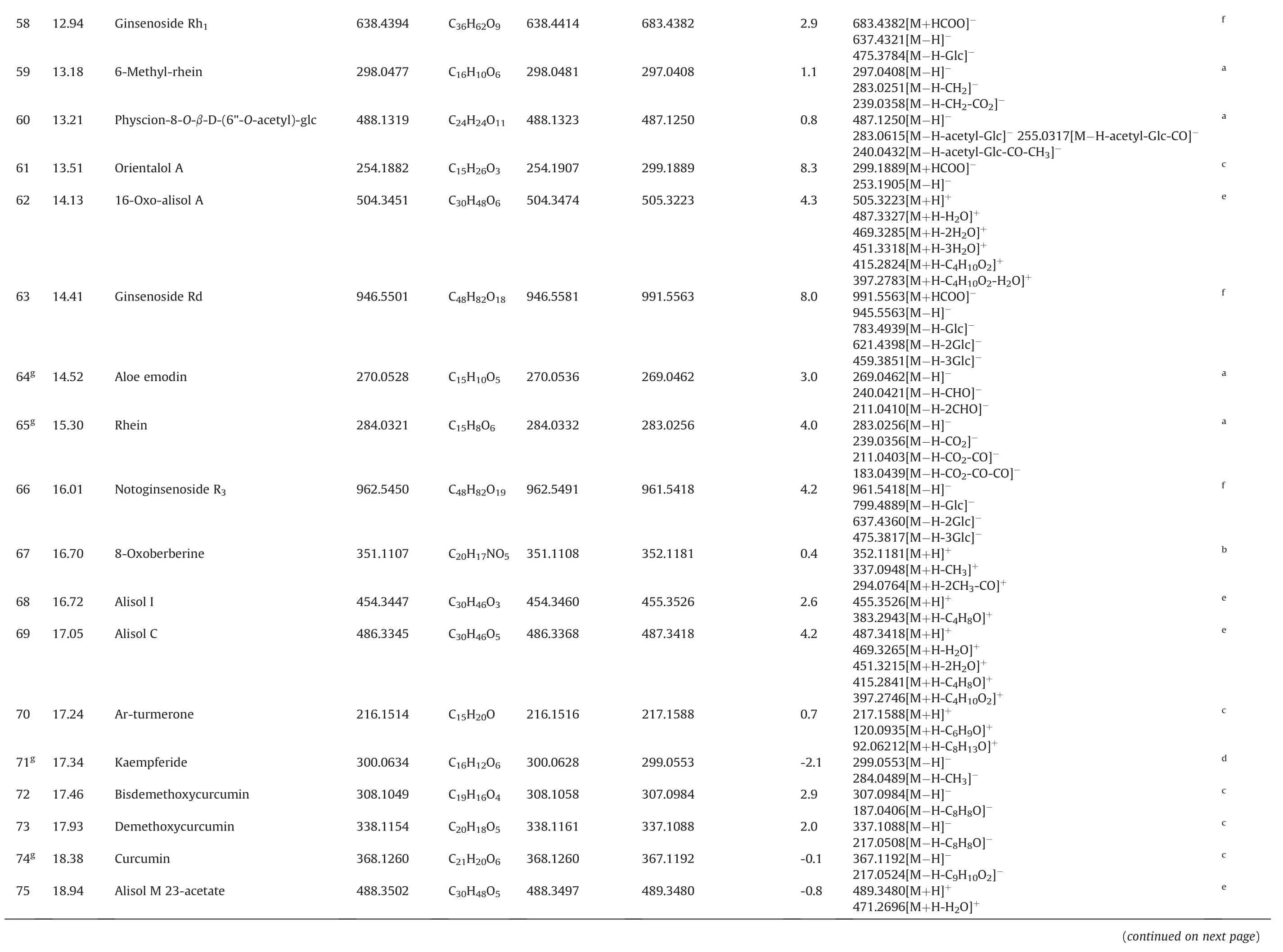
f a a c e f a a f b e e c d c c c e(continued on next page)683.4382[M+HCOO]-637.4321[M-H]-475.3784[M-H-Glc]-297.0408[M-H]-283.0251[M-H-CH2]-239.0358[M-H-CH2-CO2]-487.1250[M-H]-283.0615[M-H-acetyl-Glc]-255.0317[M-H-acetyl-Glc-CO]-240.0432[M-H-acetyl-Glc-CO-CH3]-299.1889[M+HCOO]-253.1905[M-H]-505.3223[M+H]+487.3327[M+H-H2O]+469.3285[M+H-2H2O]+451.3318[M+H-3H2O]+415.2824[M+H-C4H10O2]+397.2783[M+H-C4H10O2-H2O]+991.5563[M+HCOO]-945.5563[M-H]-783.4939[M-H-Glc]-621.4398[M-H-2Glc]-459.3851[M-H-3Glc]-269.0462[M-H]-240.0421[M-H-CHO]-211.0410[M-H-2CHO]-283.0256[M-H]-239.0356[M-H-CO2]-211.0403[M-H-CO2-CO]-183.0439[M-H-CO2-CO-CO]-961.5418[M-H]-799.4889[M-H-Glc]-637.4360[M-H-2Glc]-475.3817[M-H-3Glc]-352.1181[M+H]+337.0948[M+H-CH3]+294.0764[M+H-2CH3-CO]+455.3526[M+H]+383.2943[M+H-C4H8O]+487.3418[M+H]+469.3265[M+H-H2O]+451.3215[M+H-2H2O]+415.2841[M+H-C4H8O]+397.2746[M+H-C4H10O2]+217.1588[M+H]+120.0935[M+H-C6H9O]+92.06212[M+H-C8H13O]+299.0553[M-H]-284.0489[M-H-CH3]-307.0984[M-H]-187.0406[M-H-C8H8O]-337.1088[M-H]-217.0508[M-H-C8H8O]-367.1192[M-H]-217.0524[M-H-C9H10O2]-489.3480[M+H]+471.2696[M+H-H2O]+2.9 1.1 0.8 8.3 4.3 8.0 3.0 4.0 4.2 0.4 2.6 4.2 0.7-2.1 2.9 2.0-0.1-0.8 683.4382 297.0408 487.1250 299.1889 505.3223 991.5563 269.0462 283.0256 961.5418 352.1181 455.3526 487.3418 217.1588 299.0553 307.0984 337.1088 367.1192 489.3480 638.4414 298.0481 488.1323 254.1907 504.3474 946.5581 270.0536 284.0332 962.5491 454.3460 486.3368 216.1516 300.0628 308.1058 338.1161 368.1260 488.3497 C36H62O9 C16H10O6 C24H24O11 C15H26O3 C30H48O6 C48H82O18 C15H10O5 C15H8O6 C48H82O19 C20H17NO5 351.1108 C30H46O3 C30H46O5 C15H20O C16H12O6 C19H16O4 C20H18O5 C21H20O6 C30H48O5 638.4394 298.0477 488.1319 254.1882 504.3451 946.5501 270.0528 284.0321 962.5450 351.1107 454.3447 486.3345 216.1514 300.0634 308.1049 338.1154 368.1260 488.3502 Ginsenoside Rh1 6-Methyl-rhein Physcion-8-O-β-D-(6"-O-acetyl)-glc Orientalol A 16-Oxo-alisol A Ginsenoside Rd Aloe emodin Rhein Notoginsenoside R3 8-Oxoberberine Alisol I Alisol C Ar-turmerone Kaempferide Bisdemethoxycurcumin Demethoxycurcumin Curcumin Alisol M 23-acetate 12.94 13.18 13.21 13.51 14.13 14.41 14.52 15.30 16.01 16.70 16.72 17.05 17.24 17.34 17.46 17.93 18.38 18.94 58 59 60 61 62 63 64g 65g 66 67 68 69 70 71g 72 73 74g 75

Table 1(continued)

e 537.3554[M+Na]+515.3735[M+H]+497.3629[M+H-H2O]+479.3508[M+H-2H2O]+437.3415[M+H-H2O-HAc]+383.2688[M+H-C6H12O3]+0.5 537.3554 514.3661 C32H50O5 514.3658 Alisol B 23-acetate 25.38 96g Glc:glucose;Rha:rhamnose;Xyl:xylose;Res:resveratrol.a Rheum palmatum L.b Coptis chinensis Franch.c Curcuma longa L.f Panax notoginseng(Burk.)F.H.Chen.e Alisma orientale(Sam.)Juzep.d Lysimachia christinae Hance.g Identif ied by comparison with reference standards.
3.2.3.Protostane triterpenoids
17 protostane triterpenoids in GDD originated from Alisma orientale(Sam.)Juzep.Protostane triterpenoids showed[M+H]+ion,adduct[M+Na]+ion in positive ion mode and all possess a tetracyclic carbon skeleton.During the collision-induced dissociation(CID)process,the hydrogen rearrangement at C-23-OH resulting in C-23-C-24 bond dissociation was proposed as a characteristic CID fragmentation pathway,which can be used to further distinguish certain positional isomers containing the acetyl unit at the C-23 or C-24 position[23,24].Such compounds usually occurred successive losses of H2O,acetic acid group(HAc,60 Da)and other complex groups such as C4H8O(72 Da),C4H10O2(90 Da)and C6H12O3(132 Da).Peaks 96 was exactly identified as alisol B 23-acetate based on retention time and fragment behavior of reference standard.The high energy MSEspectra and the proposed fragment pathway of alisol B 23-acetate are depicted in Fig.5.Alisol B 23-acetate showed[M+H]+and[M+Na]+ions at m/z 515.3735 and 537.3554,which underwent several dehydrations or deacetylations to form fragment ions at m/z 497.3629[M+H-H2O]+,479.3508[M+H-2H2O]+,and 437.3415[M+H-H2O-HAc]+,and then dissociation of the C-23-C-24 bond and loss of H2O gave rise to[M+H-C6H12O3]+ion at m/z 383.2688.Peaks 87,91,and 95 showed the similar fragmentation behavior to alisol B 23-acetate,and were identified as alisol A 23-actetate,alisol B,and alisol A 24-actetate,respectively.
Peak 92 had a protonated ion[M+H]+at m/z 469.3310 with a molecular formula of C30H44O4,and formed characteristic ions at 451.3188[M+H-H2O]+and m/z 397.2745[M+H-C4H8O]+through 23-OH dehydration and C-23-C-24 dissociation.Thus,it was assigned as Alisol L.Owing to similar cleavage patterns by loss of C4H8O,peaks 69 and 88 were deduced to be alisol C and 11-deoxy-Alisol C,respectively.Peak 62 exhibited[M+H]+ion at m/z 505.3223.Three typical dehydration ions at m/z 487.3327,469.3285 and 451.3318 were generated from the hydroxyl groups.The dissociation of the C-23-C-24 bond via hydrogen rearrangement at C-23-OH produced diagnostic ion at m/z 415.2824[M+HC4H10O2]+,with further loss of H2O generating an ion at m/z 397.2783[M+H-C4H10O2-H2O]+,so peak 62 was tentatively identified as 16-oxo-alisol A.Peak 90,14Da less than that of 16-oxoalisol A and similar to 16-oxo-alisol A,was further confirmed as alisol A.
3.2.4.Flavonoids
Ten flavones and their glycosides have been screened and identified in GDD using the UNIFI workflow.It is well known that the main MS behavior of flavone aglycones was RDA fragmentation pathway and losses of small molecules and/or radicals like CH3,CO and CO2[25].For flavones glycosides,the cleavage at glycosidic linkages could happen in both positive and negative ion modes,and 162 Da(Glc),146 Da(Rha)and 308 Da(rutinoside)were the characteristic neutral loss of flavonoid-O-glycosides.The fragment ions with low m/z were the same as that of their aglycones.Among them,peaks 43 and 71 were ascertained to be quercetin and kaempferide by contrast with reference standards.Here we took quercetin and kaempferide as examples to describe the fragment patterns of these components.Quercetin displayed a deprotonated ion at m/z 301.0360 with a molecular formula of C15H10O7,and the ions at m/z 151.0451[M-H-C8H6O3]-,121.0431[M-H-C8H6O3-CH2O]-and 107.0162[M-H-C8H6O3-CH2O-CH2]-resulted from RDA cleavage.Kaempferide,with the parent ion[M-H]-at m/z 299.0553,exhibited a diagnostic ion[M-H-CH3]-at m/z 284.0489 and RDA cleavage ion at m/z 151.0055.
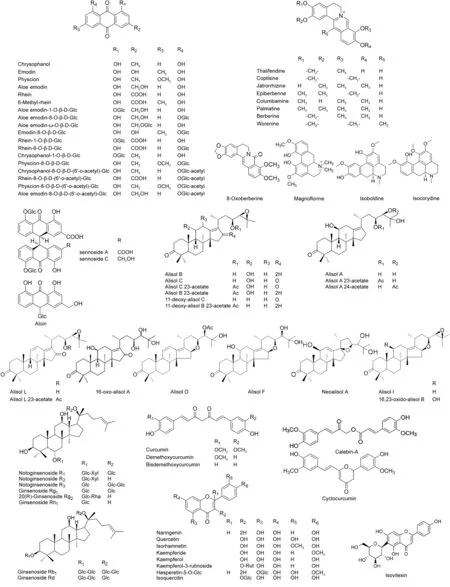
Fig.2.Chemical structures of compounds identified in GDD.

Fig.3.The MS spectra and fragmentation pathway of rhein in negative ion mode.
Peak 42 presented[M-H]-ion at m/z 285.0406,which was 14 Da less than that of kaempferide,showing the similar fragment pathways as kaempferide.It was presumed as kaempferol.Peak 23 displayed[M-H]-ion at m/z 593.1521 and produced predominant fragment ions at 447.1591[M-H-Rha]-,285.0832[M-H-Rha-Glc]-due to successive losses of glycoside fragments.Meanwhile,the ion at m/z 285.0832 further generated the characteristic ions identical to those of kaempferol,so the structure of this compound was considered as kaempferol-3-O-rutinoside.Analogously,peaks 16 and 30 were identified as rutin and hesperetin-5-O-glc,respectively.
3.2.5.Triterpenoid saponins
Eight triterpenoid saponins were detected from Panax notoginseng(Burk.)F.H.Chen in negative ion mode.These compounds offered the intense deprotonated ion[M-H]-and adduct ion[M+HCOO]-.The primary fragmentation pattern of triterpenoid saponins was the successive losses of glycosidic unit at the site of C-20,C-3 or C-6 of ginsenosides until the formation of[Aglycon-H]-ions.The species and amount of glycosyl groups were observed from MS data,in which the mass differences of 162Da,132Da and 146Da indicated the presence of glucose(Glc),xylose(Xyl),and rhamnose(Rha),respectively[26].Peak 37 was definitely identified as notoginsenoside R1with a reference standard.To facilitate characterization of these ginsenosides,the MS fragmentation pattern of notoginsenoside R1is investigated in detail(Fig.6).Notoginsenoside R1gave[M-H]-ion at m/z 931.5297 and[M+HCOO]-ion at m/z 977.5354,along with three major fragment ions at m/z 799.4856[M-H-Xyl]-,637.4314[M-H-Xyl-Glc]-,and 475.3782[M-H-Xyl-2Glc]-observed in high energy MSE spectra.Peak 55 showed deprotonated ion[M-H]-at m/z 1107.5967 with a molecular formula of C54H92O23.The fragment ion at m/z 459.3824[M-H-4Glc]-represented glycosidic cleavage by loss of four glucose residues.Hence,it was tentatively characterized as ginsenoside Rb1.
Peak 41 displayed[M-H]-and[M+HCOO]-ions at m/z 799.4863 and 845.4921,respectively,and produced fragment ion at m/z 475.3784 by loss of two glucose residues.Thus,it was tentatively assigned to be ginsenoside Rg1.Peak 54 showed deprotonated ion at m/z 769.4768.The fragment ions at m/z 637.4335[M-H-Xyl]-and 475.3786[M-H-Xyl-Glc]-corresponded to successive neutral losses of xylose residue and glucose residue,indicating that peak 54 was notoginsenoside R2.Peak 57 gave[M-H]-at m/z 783.4922,which further fragmented into m/z 637.4326[M-H-Rha]-and m/z 475.3789[M-H-Rha-Glc]-,so their fragment ions suggested that it was ginsenoside Rg2.According to the cleavage of glycosidic linkages discussed above,peaks 58,63 and 66 were tentatively identified as ginsenoside Rh1,ginsenoside Rd,and notoginsenoside R3,respectively.

Fig.4.The MS spectra and fragmentation pathway of berberine in positive ion mode.

Fig.5.The MS spectra and fragmentation pathway of alisol B 23-acetate in positive ion mode.

Fig.6.The MS spectra and fragmentation pathway of notoginsenoside R1 in negative ion mode.
3.2.6.Others
Four curcuminoids were recognized as the major active components in Curcuma longa L.Peak 74 was unambiguously identified as curcumin by comparison with a reference standard.Curcumin was taken as an example,which gave precursor ion at m/z 367.1192[M-H]-and diagnostic ion at m/z 217.0524[M-H-C9H10O2]-in negative ion mode.Analogously,peaks 56,72 and 73 were identified as cyclocurcumin,bisdemethoxycurcumin and demethoxycurcumin,respectively.The fragmentation behaviors were in accordance with those previously reported in the literature[27].
In addition,there are small amounts of tannins in GDD,of which 10 components were identified as tannins or acylglucosides by comparing with the data in library.Peak 2 showed[M-H]-ion at m/z 169.0141 as base peak,and fragment ion of m/z 125.0244 corresponding to the loss of CO2residues.So it was tentatively identified as gallic acid.Peak 5 gave[M-H]-ion at m/z 577.1359 with a molecule formula of C30H26O12,which yielded characteristic ions at m/z 425.0873[M-H-C7H4O4]-,407.0763[M-H-C7H4O4-H2O]-and 289.0712[M-H-catechin]-.Thus,it was tentatively deduced to be procyanidin B.Peak 9 exhibited[M-H]-ion at m/z 729.1471 and fragment ion at m/z 577.1350[M-H-gallate]-,suggesting that it had an additional gallate than procyanidin B.It was tentatively identified as procyanidin B-O-gallate.Based on the similar fragment pattern,peaks 1,4 and 19 were determined as gallic acid-Oglc,(+)-catechin-5-O-β-D-glc and(+)-catechin-3-O-gallate by matching the data in library,respectively.
3.3.Contribution of individual herbs to GDD
The established method was subsequently applied to analyze individual herbal decoctions by UPLC-Q-TOF-MSE,and the relative sources of 96 compounds were also correspondingly confirmed.In summary,37 components were from Rheum palmatum L.,15 components were from Coptis chinensis Franch.,9 components came from Curcuma longa L.,15 components were from Lysimachia christinae Hance,17 components were from Alisma orientale(Sam.)Juzep.and 8 triterpenoid saponins were from Panax notoginseng(Burk.)F.H.Chen.The BPI chromatograms of six individual herbs in positive and negative ion modes are shown in Fig.7.But each individual herb undoubtedly contributed to chemical components in GDD.Therefore,different sources and multiple types of pharmacodynamic components can exert better therapeutic effect through synergism or complementation.
4.Conclusion

Fig.7.The base peak intensity(BPI)chromatograms of six individual herbs in positive(A)and negative(B)ion modes.DH=Rheum palmatum L.,HL=Coptis chinensis Franch.,JH=Curcuma longa L.,JQC=Lysimachia christinae Hance,ZX=Alisma orientale(Sam.)Juzep.and SQ=Panax notoginseng(Burk.)F.H.Chen.
In this study,an integrative strategy based on UPLC-Q-TOF-MSEcoupled with UNIFI informatics platform was applied for chemical profile analysis of GDD.To the best of our knowledge,it was the first time to reveal the constituents in GDD comprehensively.By comparison with retention time,accurate mass,fragmentation behavior,a total of 96 compounds were identified or tentatively characterized from GDD,including anthraquinones,alkaloids,protostane triterpenoids,flavonoids,triterpenoid saponins,tannins,curcuminoids and other compounds.Additionally,the ESI-MS fragmentation patterns of representative compounds in different chemical structure types were investigated.Most of the high response constitutions in individual herbs were also detected in GDD.This approach provided a rapid method for high throughput screening and characterization of constituents,and would be available in other TCM formulae analysis.What is more,the results could supply valuable information for the quality control and further study of GDD in vivo.
Moreover,we found that most of the compounds have abundant phenolic hydroxyl,especially the anthraquinones,curcumin and flavonoids.The structures of these compounds tend to be easily chelated by copper ions.Therefore,it is speculated that natural small molecules in GDD that could selectively chelate copper are able to form stable complexes to promote copper excretion.These molecules with properties would serve as a promising alternative to current treatments.This work has great guiding significance in further research and application of GDD in clinical treatment.
杂志排行

Journal of Pharmaceutical Analysis的其它文章
- Karacoline,identified by network pharmacology,reduces degradation of the extracellular matrix in intervertebral disc degeneration via the NF-κB signaling pathway
- Nanodiamonds with powerful ability for drug delivery and biomedical applications:Recent updates on in vivo study and patents
- Comparing different domains of analysis for the characterisation of N-glycans on monoclonal antibodies
- GC-NICI-MS analysis of acetazolamide and other sulfonamide(R-SO2-NH2)drugs as pentafluorobenzyl derivatives[R-SO2-N(PFB)2]and quantification of pharmacological acetazolamide in human urine
- Analysis of pesticide residues in commercially available chenpi using a modified QuEChERS method and GC-MS/MS determination
- Determination of L-norvaline and L-tryptophan in dietary supplements by nano-LC using an O-[2-(methacryloyloxy)-ethylcarbamoyl]-10,11-dihydroquinidine-silica hybrid monolithic column