Prevalence and genotype distribution of hepatitis B virus among migrant workers in Lombok Island, Indonesia
2020-02-27LauraNavikaYamaniEvaTrianiMochamadAminJuniastutiTakakoUtsumiSoetjiptoNasronudinYoshihikoYanoHakHottaYoshitakeHayashiMariaIngeLusida
Laura Navika Yamani, Eva Triani, Mochamad Amin, Juniastuti,5, Takako Utsumi, Soetjipto,7, Nasronudin, Yoshihiko Yano, Hak Hotta, Yoshitake Hayashi, Maria Inge Lusida,5✉
1Institute of Tropical Disease, Universitas Airlangga, Surabaya, East Java 60115, Indonesia
2Department of Epidemiology, Faculty of Public Health, Universitas Airlangga, Surabaya, East Java 60115, Indonesia
3Indonesia-Japan Collaborative Research Center for Emerging and Re-emerging Infectious Diseases, Institute of Tropical Disease, Universitas
Airlangga, Surabaya, East Java 60115, Indonesia
4Faculty of Medicine, Mataram University, West Nusa Tenggara, Indonesia
5Department of Microbiology, Faculty of Medicine, Universitas Airlangga, Surabaya, East Java 60131, Indonesia
6Center for Infectious Diseases, Kobe University Graduate School of Medicine, Kobe, Hyogo 6500017, Japan
7Department of Biochemistry, Faculty of Medicine, Universitas Airlangga, Surabaya, East Java 60131, Indonesia
ABSTRACT Objective: To examine the potential risk of hepatitis B virus (HBV) spread in Indonesia by migrant workers, based on the molecular characteristics of HBV strains.Methods: Sera collected from migrant workers traveling to their destination countries (pre-migrant workers) and those returning to Indonesia (post-migrant workers) were screened for HBsAg by ELISA, followed by HBV DNA detection by PCR and (sub)genotype/subtype determination according to surface region and whole genome sequencing. Results: Of 87 pre-migrant workers, 15 (17.24%) were HBsAgpositive, whereas 15 (12.10%) of 124 post-migrant workers were HBsAg seropositive. HBV genotype analysis based on the S region showed that HBV-B3/adw2 was predominant (96.15%, 25/26) whereas 3.85% (1/26) of isolates were HBV-C3/adrq+. Whole genome sequencing of selected strains and phylogenetic tree analysis identified subgenotype B7 in three samples previously categorized as subgenotype B3 based on S region analysis, supporting a recent argument that subgenotypes B5/B7/B8/B9 could be considered as a quasi-subgenotype of B3. Conclusions: A high prevalence of HBsAg carriers was detected among migrant workers from Lombok Island, with no significant difference in prevalence between before and after returning to Indonesia. All strains were classified into genotypes common in Indonesia, and the results suggested that migrant workers are not a risk factor for HBV transmission into Indonesia.
KEYWORDS: HBV; Subgenotypes; Migrant workers; Indonesia
1. Introduction
Among an estimated 257 million people with hepatitis B virus (HBV) infection worldwide, more than 50% live in Asian countries. The prevalence of chronic HBV infection varies widely in different parts of the world, and regions are categorized into high (>8%), intermediate (2%-8%), and low (<2%) endemicity. HBV is classified into different genotypes based on intergenotypic divergence of at least 8% in the complete genome or 4% in the surface (S) gene[1-3]. HBV is classified into nine genotypes (A-I) and one ‘putative’ genotype (J) based on divergence over the complete genome[4-9]. In addition, nine serological types, referred to as subtypes adw2, adw4, adrq+, adrq-, ayw1, ayw2, ayw3, ayw4, and ayr have been reported based on the different antigenic determinants of the HBsAg surface antigen[10,11]. According to the World Health Organization, there is a high prevalence of HBV infection in Southeast Asian countries, among which Indonesia has a moderate to high endemicity. The distribution of HBV genotypes among HBV-infected individuals in various islands of Indonesia, including Lombok, is ethnic- and geographic-specific[12-14]. The largest ethnic group in Lombok Island is the Sasak tribe. The majority of migrant workers from Indonesia originate from Lombok Island, and most of the inhabitants of migrant workers from Lombok work as farm laborers in Malaysia, Saudi Arabia, Brunei, Taiwan (China), and Korea. Lombok’s large population of migrant workers may have a negative impact on the prevalence of HBV infection in Indonesia because the mobility, migration, and occupational instability of migrant workers may increase their opportunities to expose with hepatitis B carriers[15]. The recent trend toward increased mobility and globalization may accelerate the spread of HBV and change the molecular epidemiological pattern of HBV. Therefore, determining the prevalence of HBV infection among migrant workers, especially in Lombok Island, is important. The present study examined the potential risk of HBV spread in Indonesia by migrant workers, based on the molecular characteristics of HBV strains. We analyzed the prevalence of HBV infection among Indonesian migrant workers, including pre-migrant (migrant workers who will go to their destination countries for the first time) and post-migrant (those returning to Indonesia) workers. In addition, the genotypes/subgenotypes of HBV were analyzed based on surface region and whole genome HBV sequences to determine the distribution of HBV genotypes/subgenotypes among Indonesian migrant workers. Infected pre- and post-migrant workers were analyzed to determine whether the HBV infection was acquired in Indonesia or in their destination countries. Improving our knowledge of HBV infection among migrant workers could be beneficial for preventing and controlling the spread of HBV.
2. Materials and methods
2.1. Sample collection and ethical approval
A cross-sectional, descriptive study was carried out among 211 Indonesian migrant workers. However, the number of samples was designed by the calculation in 5% precision and confidence level 95%, showing the minimum 196 samples needed.
Serum samples were collected in 2013 from Indonesian migrant workers from the two study groups, which included 87 pre-migrant workers (52 women and 35 men), and 124 post-migrant workers (66 women and 58 men). A total of 211 serum samples were collected from Indonesian migrant workers domiciled in Lombok Island who presented to Hepatika Laboratory Mataram for health checks and were screened for HBsAg. All participants were tested for hepatitis B surface antigen (HBsAg) using a commercial ELISA kit (Hepalisa kit, J. Mitra & Co. Pvt. Ltd., New Delhi, India). The study was approved by the ethical committee of Universitas Airlangga (No. 61/UN18.8/ETIK/2013). Informed consent has been obtained from all individuals included in this study.
2.2. HBV DNA extraction and PCR amplification
HBV DNA was extracted from 200 µL of HBsAg positive sera using a QIAamp DNA Blood Mini Kit (Qiagen, Tokyo, Japan) following the manufacturer’s instructions. The HBV S gene (nt 256-796) was detected using first-step PCR with primers P7 and P8 as described previously[16]. Nested PCR was performed in case of negative first-step PCR results. The second-step PCR amplified 258 bp of the S gene (nt 455-713) with primers HBS1 and HBS2 as described previously[17] and using the first-step PCR product as the template. Complete HBV genome sequencing was performed using a previously reported method[8,18]. In brief, the complete HBV genome was first amplified as two overlapping fragments, a 3 200-bp amplicon (Fragment A) and a 462-bp amplicon (Fragment B) covering the remaining region. Part A was then subjected to nested PCR to amplify 11 overlapping fragments as described previously[18]. The HBV DNA amplicons were separated using 2% agarose gel electrophoresis at 100 V for 35 min and stained with ethidium bromide. PCR product size was estimated in comparison with a 100-bp DNA ladder under UV light.
2.3. Sequencing and phylogenetic analysis
The PCR products were purified using the QIA quick PCR purification Kit (Qiagen Hilden, Germany) according to the manufacturer’s instructions. The sequencing reactions were prepared using the Big Dye Deoxy Terminator cycle sequencing kit V3.1 with an ABI Prism 3500XL genetic analyzer (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions. Reference sequences were retrieved from the DDBJ/EMBL/GenBank databases. Alignments were performed using CLUSTAL X, and the phylogenetic trees were constructed using the neighborjoining method[16]. Bootstrap values were determined on 1 000 resamplings of the datasets[19]. The final tree was obtained using MEGA6 software (http://www.megasoftware.net/). HBV genotype and subgenotypes were determined according to the results of phylogenetic analysis.
2.4. Nucleotide sequence accession numbers
The nucleotide sequence data of the HBV whole genome reported in this paper will appear in the DDBJ/EMBL/GenBank databases under accession No. LC416036 through LC416040.
2.5. Statistical analysis
The positivity for HBsAg, HBV-DNA, HBV sub/genotypes were described using proportions. The SPSS software program, version 7, was used. Ninety-five percent confidence intervals (CIs) for the proportion of individuals who were HBsAg-positive and HBV-DNA positive were calculated for each variable.
3. Results
The study included 87 (41.23%) pre-migrant workers and 124 (58.77%) post-migrant workers who had worked abroad and planned to return to work overseas. The number of Indonesian HBsAgpositive pre- and post-migrant workers were 15/87 (17.24%) and 15/124 (12.10%), respectively. Thirty HBsAg-positive serum samples (10 men and 20 women; age range: 20-50 years) were included in the molecular epidemiology study. All HBsAg-positive samples were HBV DNA positive. Of the 30 samples, 22 (73.33%) were positive in the first-step PCR using the P7-P8 primers (540 bp), 8 (26.67%) were positive in the second-step PCR using the HBS1 and HBS2 primers (258 bp), and 4 had poor results and were excluded from subsequent analysis. Accordingly, 26 samples were analyzed by S gene sequencing (Table 1).
The HBsAg positive in pre-and post-migrant workers were found 17.24% (95% CI: 10.28%-27.16%) and 12.10% (95% CI: 7.16%-19.47%), respectively. While HBV DNA identified as PCR positive results were detected 12.64% (95% CI: 6.78%-21.91%) and 12.10% (95% CI: 7.16%-19.47%) in pre- and post-migrant workers, respectively. Overall, HBsAg and PCR positive results obtained in migrant workers were 14.22% (95% CI: 9.95%-19.84%) and 12.32% (95% CI: 8.35%-17.71%), respectively.
Figures 1 and 2 show the results of phylogenetic analysis of samples and reference sequences from the GenBank database based on a partial sequence of the S gene region of the HBV genome. Almost all of the samples (25/26, 96.15%) were genotype B and one sample was genotype C (1/26, 3.85%). The phylogenetic tree showed that 21 samples amplified by primers P7-P8 belonged to subgenotype B3 and 1 sample belonged to subgenotype C3 (Figure 1). Four samples amplified by primers HBS1 and HBS2 were included in the sequencing analysis, and all belonged to subgenotype B3 (Figure 2). Of the 26 samples analyzed for HBV subtypes, 25 were identified as HBV adw2 with genotype B and 1 was identified as subtype adrq+ with genotype C.
The subgenotype results obtained by S region sequencing were reconfirmed by selecting four samples (HBV-TKI.2, HBV-TKI.11, HBV-TKI.16, and HBV-CT.13) with different branch lengths in the phylogenetic tree in the subgenotype B3 group and the only sample (HBV-CT.1) with subgenotype C3. These samples were subjected to whole genome HBV analysis using different primer sets[18]. In the phylogenetic tree analysis of whole genome HBV, one sample (HBVTKI.16) was confirmed as B3, three samples (HBV-TKI.2, HBVTKI.11, and HBV-CT.13) tended to be closer to subgenotype B7 than to B3, and one sample (HBV-CT.1) was identified as subgenotype C7 rather than C3 (Figure 3). The amino acid sequences of HBsAg from five representative Indonesian migrant workers were compared with reference sequences from HBV subgenotypes. Based on data from aa114 to aa180, the results of four HBV genotype B sequences confirmed that these were similar to those of subgenotype B3 at these positions, whereas one HBV genotype C sequence was similar to that of subgenotype C7 (Figure 4). Nucleotide sequence analysis by phylogenetic tree and amino acid sequence analysis based on the S gene in four samples (HBV-TKI.2, HBV-TKI.11, HBV-TKI.16,
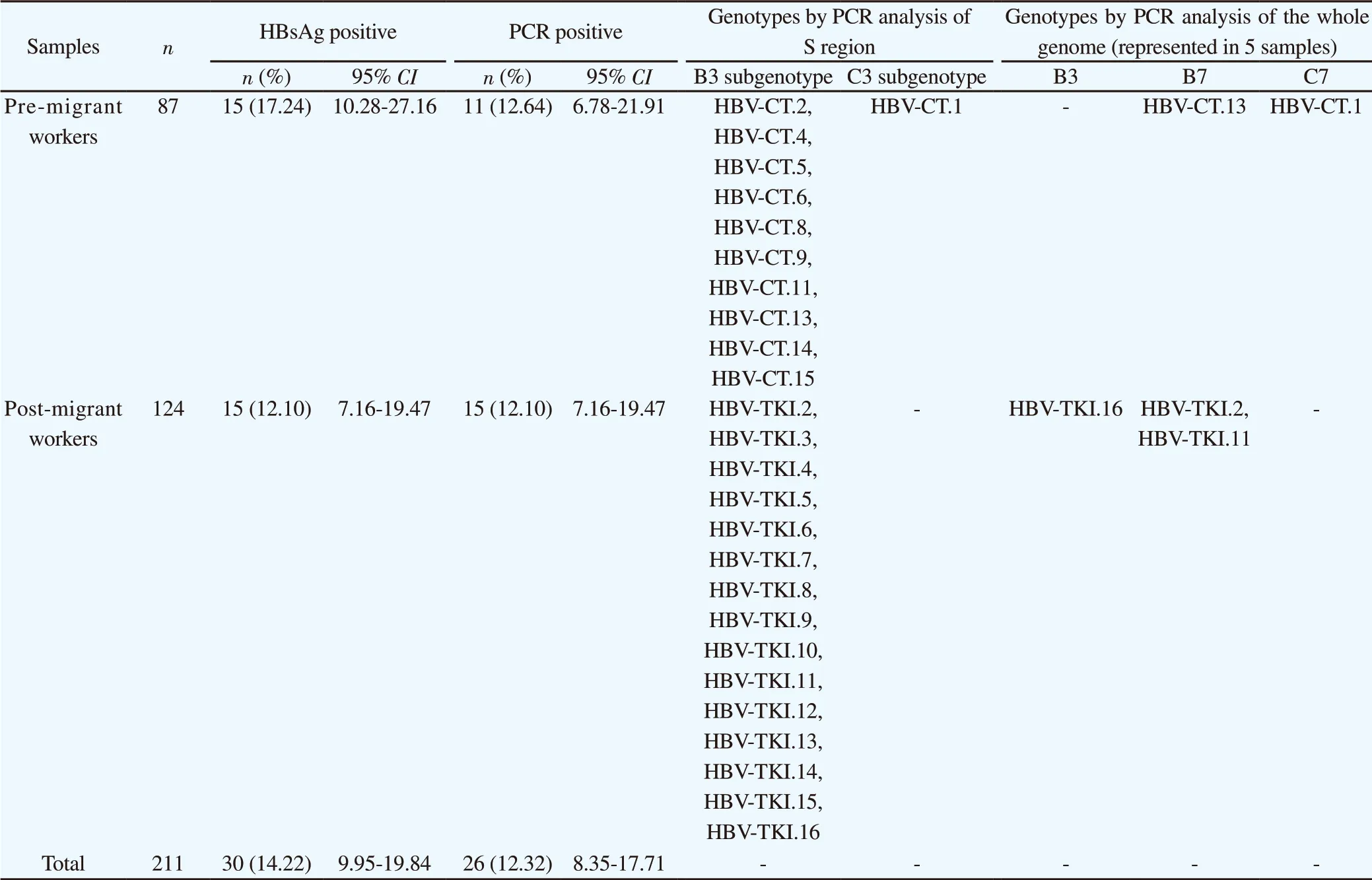
Table 1. Analysis of migrant worker samples detected by HBsAg serology, PCR in S region and whole genome between pre-migrant workers and post migrant workers.
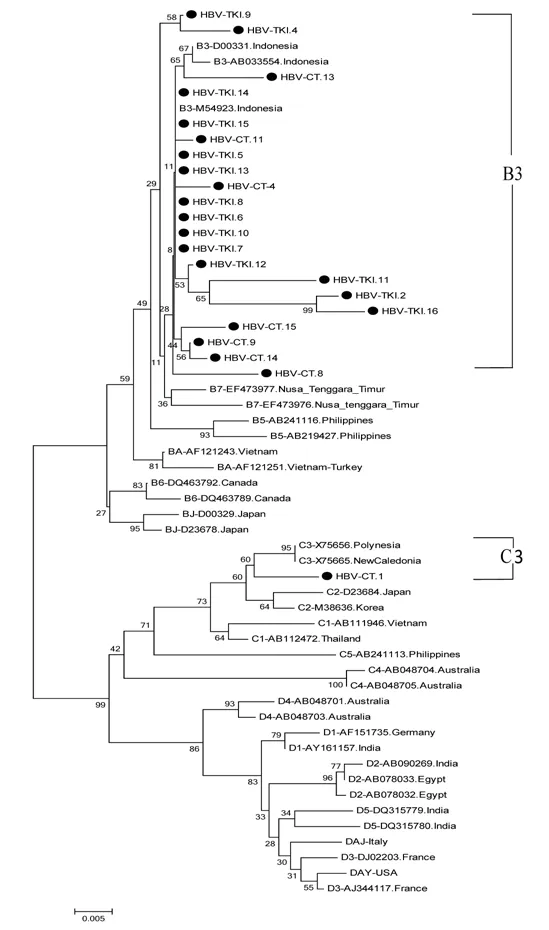
Figure 1. The phylogenetic tree of 22 HBV isolates amplified in S region by primer P7-P8 from Indonesian migrants workers domicilied in Lombok Island [code HBV-CT (pre-migrant workers) and HBV-TKI (post-migrant workers); shown as labeled], and reference sequences from GenBank databases (indicated with the accession numbers and countries of origin). Twenty two samples belonged to subgenotype B3 and 1 sample was C3 based on S region. The genotypes and subgenogroups of the samples are indicated on the branches, and displayed on the right.and HBV-CT.13) showed the same subgenotype, whereas three of the four samples appeared to belong to subgenotype B7 when analyzed using the full-length genome. The five whole genome sequences of HBV/B and the HBV/C sequence (HBV-CT.13, HBVTKI.2, HBV-TKI.11, HBV-TKI.16, and HBV-CT.1) were submitted to the International DDBJ/EMBL/GenBank Database with accession numbers LC416036 through LC416040, respectively.
4. Discussion
Migration and rapid population mobility may change the epidemiology of infectious diseases in different areas of the world[20]. Population mobility has been identified as an independent risk factor for the spread of HBV infection because the virus can be transferred to and from different locations as individuals move between regions[21]. In Indonesia, population mobility is associated with the movement of migrant workers because many Indonesians work overseas in East Asia, America, the Middle East, and Europe as domestic workers, caregivers, operators, and plantation workers. Data from the National Agency for Placement and Protection of Indonesian Workers (BNP2TKI) showed that in 2017, the five Indonesian districts or cities with the highest numbers of migrant workers were in descending order: East Lombok, Indramayu, Central Lombok, Cirebon, and Cilacap. Therefore, Lombok Island (including the districts East, Central, West, and North Lombok) contributes the highest number of migrant workers. Most of the migrant workers from Lombok work as farm laborers in Malaysia, Saudi Arabia, Brunei, Taiwan (China), and Korea. Lombok has become the largest area of origin of workers migrating to Malaysia after East Java, sending 47 000 migrant workers in 2014, 35 000 in 2015, and 18 000 in 2016. Most of these laborers work in oil palm and rubber plantations. To the best of our knowledge, the present study is the first to analyze the molecular epidemiology of HBV circulating in migrant workers domiciled in Lombok Island.
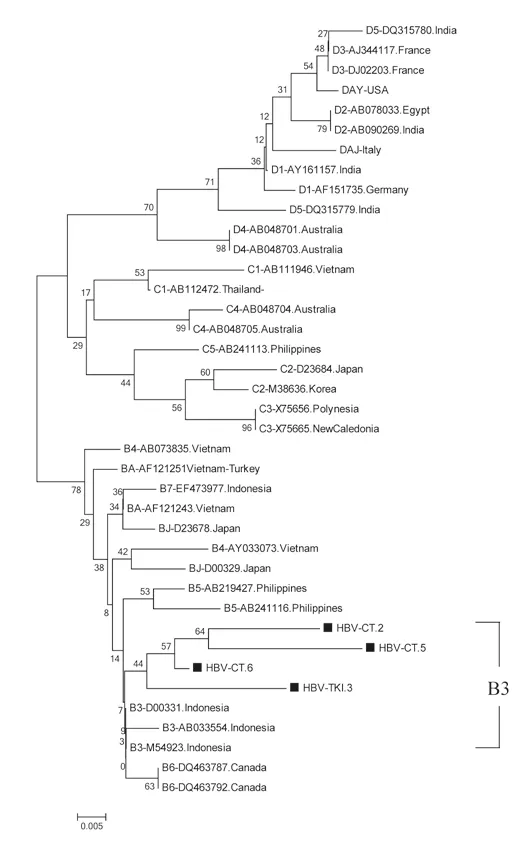
Figure 2. The phylogenetic tree of 4 HBV isolates amplified in S region by primer HBS1 and HBS2 from Indonesian migrants workers domicilied in Lombok Island [code HBV-CT (pre-migrant workers) and HBV-TKI (post-migrant workers); shown in labeled], and reference sequences from GenBank databases (indicated with the accession numbers and countries of origin). The genotypes and subgenogroups of samples are indicated on the branches, and displayed on the right.
The present study included 211 participants from Indonesia domiciled in Lombok Island including 87 pre-migrant workers and 124 post-migrant workers. Of 30 HBsAg-positive samples identified, 15 (17.24%) were from pre-migrant workers and 15 (12.10%) from post-migrant workers. The prevalence of HBsAg-positive samples was higher than that reported among migrant workers from other South East Asian countries such as Cambodia (10.8%), Laos (6.9%), and Myanmar (9.7%)[22]. A previous study reported an HBsAg positivity rate of 18.6% among 479 adults in Lombok[12], a rate higher than that for Indonesia as a whole, which is considered to have moderate-to-high hepatitis B endemicity[23]. Mass neonatal vaccination for hepatitis B was started in Indonesia only in 1997[24].
In addition, the socio-cultural patterns in Lombok communities, particularly those of the Sasak tribe which is native to Lombok, are characterized by frequent divorce and remarriage[25], thus contributing to the increased prevalence of HBV infection in these communities.
The majority of the 26 samples sequenced (25/26, 96.15%) were identified as genotype B and only 1 (3.85%) as genotype C, in agreement with a previous study that found a predominance of HBV-B over HBV-C in Lombok[12]. We did not identify HBV genotypes specific to Middle-East countries and East Asia, which are destination countries. The sequence data of the 26 samples analyzed by the S region in the phylogenetic tree revealed 25 samples with similar branches to those of subgenotype B3, whereas one sample was classified as subgenotype C3. Subgenotype B3 is the dominant type in Eastern Indonesia (where Lombok is located) and in Denpasar (Bali), followed by subgenotype B8. In HBV genotype C, subgenotype C7 was predominant, followed by C1[12]. Several studies have reported the frequent isolation of subgenotype B3 from Indonesia[12,26-31]. That subgenotype B3 is specific to Indonesia and is not found elsewhere which suggests that B3 evolved independently from its ancestor in Indonesia.
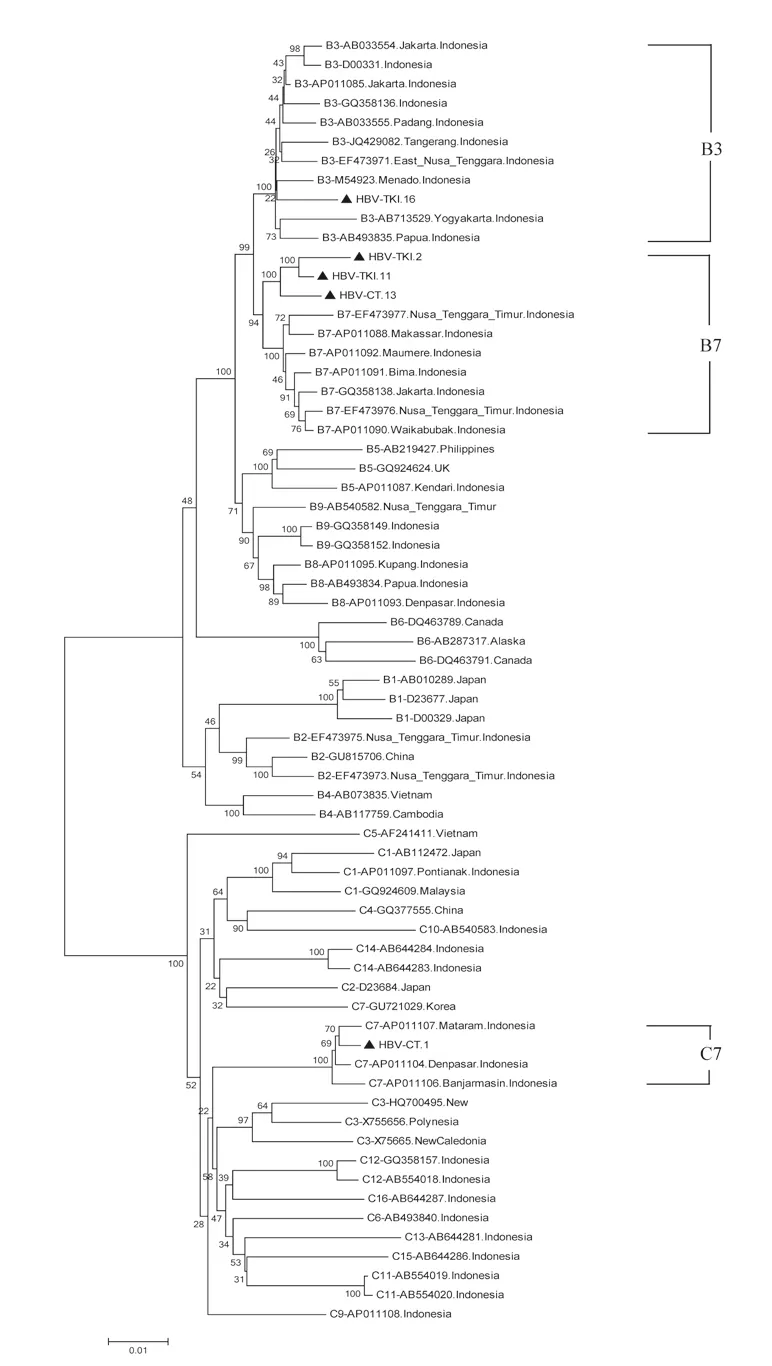
Figure 3. The full-length genome of 5 HBV isolates from Indonesian migrants workers domicilied in Lombok Island [code HBV-CT (pre-migrant workers) and HBV-TKI (post-migrant workers); shown as labeled] analyzed by phylogenetic tree and compared with reference sequences from GenBank databases (indicated with the accession numbers and countries of origin). The genotypes and subgenogroups of samples are indicated on the branches, and displayed on the right.

Figure 4. Amino acid sequences of HBsAg from five representative Indonesian migrant workers compared with reference sequences from GenBank databases at amino acid positions 114 to 180. Four samples with HBV genotype B (HBV-CT.13, HBV-TKI.2, HBV-TKI.11, and HBV-TKI.16) were similar to B1, B2, and B3, whereas one HBV genotype C (HBV-CT.1) sample showed complete homology to C1, C2, and C7.
HBV (sub) genotype determination can be made according to the S region as a replacement for whole genome analysis[14], but complete genome sequencing is more accurate for the classification of subgenotypes[13,32,33]. Moreover, whole genome sequencing provides a better understanding of the genetic variation of HBV derived from mutations or recombination that may occur in different genes. The correct classification of HBV subgenotypes within the same genotype is based on a nucleotide divergence of 4.0%-7.5% over the whole genome[33]. Studies show that using a partial genome can lead to the misclassification of HBV subgenotypes[34- 37]. The present study investigated this phenomenon, and the analysis identified different HBV subgenotypes using the nucleotide sequence of the S gene and the whole HBV genome. The phylogenetic analysis of the S region in four samples with subgenotype B3, HBVTKI.2, HBV-TKI.11, HBV-TKI.16, and HBV-CT.13 revealed that they may belong to a different group based on the long branch of the phylogenetic tree. To confirm this finding, the four samples and one sample with subgenotype C3 were analyzed by the HBV full-length genome using sets of PCR primers that amplify the whole genome. One sample (HBV-TKI.16) was definitely grouped into subgenotype B3, whereas the three other samples (HBV-TKI.2, HBV-TKI.11, and HBV-CT.13) were closer to subgenotype B7 than to subgenotype B3, and the C3 sample was changed into subgenotype C7. Such misclassification of subgenotypes, particularly for the HBV subgenotype B3, in which samples were classified as B3 by S region and B7 according to whole genome sequencing, has been reported previously[36]. Therefore, it has been proposed that HBV genotype B should be reclassified to consider subgenotypes B5, B7, B8, and B9 as quasi-subgenotypes of B3 in Southeast Asia[33]. Three of these subgenotypes (B7, B8, and B9) were identified from Indonesian isolates. HBV subgenotype B7 was first isolated from the Nusa Tenggara Islands in Eastern Indonesia in 2008[13]. Thereafter, subgenotype B8 was also found in Eastern Indonesia, in Bali, Sulawesi, and East Nusa Tenggara[12]. The most recently identified subgenotype B9 was isolated from the same region in Indonesia where subgenotype B8 was found. This indicated that the distribution of HBV subgenotypes B7, B8, and B9 is associated with the ethnic origin of Eastern Indonesia. However, subgenotype B7 is found in West Nusa Tenggara, which includes Lombok, and also in East Nusa Tenggara. The phylogenetic analysis of the full-length HBV genome in the present study identified HBV genotypes B3, B7, and C7, which originate from Indonesia.
Analysis of HBV subtypes showed that almost all of the samples (96%) had HBV subtype adw2, and one sample had the adrq+ subtype. Adw2 was the predominant subtype in the present study. The adw subtype is found in large areas in Africa, the Eastern Mediterranean region, Western Asia to Northern India, Western Europe, and North and South America[1,38]. The adw subtype is mainly detected in Southern areas such as Southern China, Okinawa and Amami, the Philippines, and Indonesia[39,40]. The HBV subtype adw2 and subtype adrq+ are found predominantly in genotype B and C in Indonesia[12,41]. The epidemiology of HBV subtypes may indicate geographic and ethnic differences in its distribution. The subtype distribution patterns may reflect previous migration patterns, as HBV infection transmitted between individuals eventually results in a homogeneous HBV subtype distribution. The HBV subtype is related to ethnic and genetic factors, particularly in individuals with chronic disease[12,23,42]. In Lombok, migrant work has increased the mobility of the population and caused demographic changes, which may have led to an obscure pattern of HBV genotype,subgenotype, and subtype distribution. In the present study, the prevalence of HBV infection was similar between pre- and postmigrant workers. In addition, the HBV (sub)genotypes and subtypes did not differ significantly between pre-migrant workers and postmigrant workers. This finding suggests that these workers were most likely to have become infected in Lombok. As there were no novel subgenotypes specific to post-migrant workers who lived overseas, migrant workers did not import HBV from other countries. Our data may be valuable for the local government/immigration to prevent the transmission of HBV infection into countries of destination. This study has limitations, because clinical data of hepatitis status was not obtained from migrant workers. Moreover, our study was limited to detect HBV in migrant workers domicilied in Lombok Island. It is suggested that the result be not representative yet about the global prevalence in Indonesia. We need further investigation to conduct HBV detection in migrant workers domicilied from other areas. This is a cross sectional study, in which samples analyzed in pre migrant workers were different from those in post migrant workers. Furthermore, we need to monitor the viral transmission during migration with cohort study.
In conclusion, the present study showed a high prevalence of HBV infection in pre- and post-migrant workers originating from Lombok Island, Indonesia, around 14.22% of total samples. Analysis of HBV (sub)genotypes among migrant workers in Lombok showed that they contracted HBV in their home island rather than in the destination country. The present study also showed that the HBV S gene sequence is not accurate for HBV subgenotype determination as it is not always representative of the whole genome.
Conflict of interest statement
No potential conflicts of interests with respect to publication of this article is stated here.
Funding
This work was partly supported by the Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (16H05826), and a Grant-in-Aid from the Japan Initiative for Global Research Network on Infectious Disease (J-GRID) supported by the Ministry of Education, Culture, Sports, Science and Technology, Japan. The present study was also supported by Grantin-Aid from Professor Dato’ Sri Tahir through Tahir professorship, Indonesia. We are also grateful to Hepatika Laboratory Mataram for providing serum samples obtained from migrant workers and University of Mataram, School of Medicine, Mataram West Nusa Tenggara.
Authors’ contributions
The concept study was conducted and designed by ET, LNY, MIL and S. MA, and ET performed the samples collection and laboratory experiments. LNY and ET were responsible for the analysis and interpretation of the data. LNY wrote and drafted the manuscript. MIL, S, J, N, TU, HH, YH, and YY reviewed and edited the manuscript, and then gave final approval of the version to be published. All author read and approved the final manuscript.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- The phytochemical and pharmacological properties of artocarpin from Artocarpus heterophyllus
- Salivary gland antigens of laboratory-bred Phlebotomus sergenti and their immunogenicity in human volunteers in laboratory condition
- Indoor spray and windows screens effects on dengue vector density after space spraying in a field trial
- Sandfly fauna and ecological analysis of Phlebotomus orientalis and Phlebotomus martini in the lowland foci of visceral leishmaniasis in Somali Regional State, southeast Ethiopia
- Orostachys japonicus ethyl acetate fraction suppresses MRSA biofilm formation
- Cryptococcal meningitis with pulmonary cryptococcoma in an immunocompetent patient: A case report