乙酸和亚硫酸钠协同处理对泡桐组分及结构特性的影响
2020-02-25贾亮亮齐学敏张军华常德龙许雅雅
马 莉, 贾亮亮, 齐学敏, 楚 杰*, 张军华, 常德龙, 许雅雅
1. 西北农林科技大学, 陕西 杨凌 712100 2. 国家林业和草原局泡桐研究开发中心, 河南 郑州 450003
引 言
生物质能被称为世界上的“第四大能源”[1]。 其中木质生物质能是可再生和可持续发展的[2]。 随着全球气候变暖以及不可再生资源的日益消耗, 木质生物质能的有效利用越来越受到工业应用的关注[3]。 近年来世界能源工业不断发展, 越来越多的木质生物质材料(如能源草, 农作物秸秆, 木材废料等)被用作生产工业乙醇的绿色生物质材料。
泡桐(Paulownia)是一种生长周期短、 多用途树种, 可用于制浆、 家具、 手工艺品、 乐器和木炭等领域。 泡桐原产于中国, 年产量极高, 约50 t·a-1, 显著高于其他能源物种(如杨树, 柳枝稷, 芒草及柳树等)的产量约6~17 t·a-1 [4]。 此外, 我国泡桐资源丰富, 种植面积约968万亩, 约占世界种植面积的27%。 在当前泡桐产业发展中, 主要以桐木单板和桐木家具加工为主[5], 桐木综合利用价值较低, 加工生产过程中会产生大量的加工废弃物。 因此, 有效利用这些废弃泡桐资源进行生物质能源转化来制备燃料乙醇是非常有意义的。
有研究报道, 泡桐富含木质纤维素, 其综纤维素含量约占60%~70%[6]。 与其他木质生物质材料相比, 泡桐所含灰分和可溶性物质较少, 结构较为疏松, 是制备生物乙醇及其他化学品极具潜力的原料。 木质素和半纤维素, 尤其是木质素, 被认为是限制纤维素酶对纤维素可及性的最大物理屏障[7]。 复杂顽抗的木质素结构在很大程度上限制了泡桐的有效利用, 因此需要先对原料进行预处理破坏其结构, 增加纤维素酶与纤维素的特异性吸附, 使纤维素易于水解, 高效转化为燃料乙醇。
目前国内以泡桐作为材料制备燃料乙醇的研究较少。 部分学者使用稀酸法、 水热法、 物理化学法等对泡桐进行处理。 但针对乙酸及亚硫酸钠协同处理泡桐的研究鲜见, 乙酸预处理可以较好地降解木聚糖, 亚硫酸钠具有很好的脱木素作用。 本工作以速生泡桐为研究对象, 采用乙酸协同亚硫酸钠对原料进行化学预处理。 探究不同处理条件对泡桐化学组分及结构特性的影响, 为我国泡桐生物质资源高效转化利用提供部分理论基础。
1 实验部分
1.1 材料与仪器
泡桐木粉(加工废料), 粉碎研磨至80目, 晾干装袋贮存备用, 含水率保持在8%以下。 冰醋酸、 无水乙醇、 氢氧化钠、 无水亚硫酸钠、 浓硫酸、 葡萄糖、 木糖等均为市售分析纯试剂。
水浴锅、 电子天平、 真空泵、 干燥箱、 高速离心机、 紫外-可见分光光度计、 振荡器、 高效液相色谱、 高压灭菌锅、 高温油浴锅、 粉碎机等。
1.2 方法
1.2.1 预处理
乙酸预处理: 配置5% CH3COOH溶液250 mL, 称取绝干泡桐粉末20 g, 料液比为1∶10, 170 ℃条件下处理30 min。 结束后冷却过滤, 将固体用蒸馏水洗至中性, 晾干装袋备用。 残渣标记为RMA。
亚硫酸钠协同处理: 配置4% Na2SO3溶液250 mL, 称取RMA绝干原料20 g, 料液比为1∶10, 120 ℃条件下处理60 min, 后续处理同上。 此残渣标记为RMAS。
碱性亚硫酸钠协同处理: 配置4% Na2SO3+1% NaOH溶液250 mL, 称取RMA绝干原料20 g, 料液比为1∶10, 120 ℃条件下处理60 min, 后续处理同上。 此残渣标记为RMAAS。
1.2.2 化学组分测定
泡桐样品各组分含量参照美国国家可再生能源实验室(NREL)的方法进行分析测定。 葡萄糖及木糖峰面积在高效液相色谱仪(Agilent1200)上用Bio-Rad HPX-87H色谱柱测定。 所有试验均采用三组平行, 最终数据取平均值并计算标准误差。
1.2.3 结构表征
X-射线衍射仪(Rigaku D/max-3C generator)用于测定泡桐样品的纤维素结晶度, 测定电压40 000 V, 电流0.1 A, 扫描角度2θ, 范围为5°~50°, 扫描速率为12°·h-1。 纤维素的结晶度指数由X射线衍射仪测得的光谱峰高进行公式计算得到[8]。I002为002晶面的极大衍射强度, 2θ约为22°;Iam为非结晶背景衍射的散射强度, 2θ约为18°。 结晶度(CI)计算公式为式(1)
(1)
采用NicoletiS10型傅里叶变换红外光谱仪对样品进行红外分析。 扫描范围取4 000~400 cm-1, 分辨率为4 cm-1, 扫描次数为64次。
X射线光电子能谱仪(XPS)用于检测泡桐样品表层(2~10 nm)的原子组成及化学环境[8]。 使用的光源为单色光Al Kα X-射线源(15 kV, 5 mA)。
2 结果与讨论
2.1 预处理对泡桐组分的影响
预处理前后泡桐样品的化学组分变化如表1所示。

表1 不同预处理方法对样品化学组分及结晶度的影响
Note: RM: Raw material; RMA: Raw material+Acetic acid; RMAS: Raw material+Acetic acid+Sodium sulfite; RMAAS: Raw material+Acetic acid+Alkaline Sodium sulfite
未处理泡桐的葡聚糖, 木聚糖和木质素含量分别为46.81%, 18.92%和26.40%, 抽提物含量为5%左右。 经乙酸预处理后, 木聚糖含量降低至11.06%, 葡聚糖含量增加至50.23%。 可能是由于乙酸对木聚糖具有很好的降解溶出作用, 此结果与前人的研究相似[9]。 Wang等[9]提出160 ℃条件下, 用25%的乙酸溶液处理10%固体浓度的麦草几乎可以移除所有的木聚糖并回收94.2%的葡聚糖。 乙酸处理过程中, 木聚糖在水合氢离子的作用下, 降解为低聚木糖溶于上清液中, 使更多的纤维素暴露在预处理残渣中。 然而, 乙酸预处理对木质素的脱除十分有限, 因此三大素之间的复杂交联结构需要进一步被破坏以提高后续酶水解效率。
经亚硫酸钠协同处理后, 葡聚糖含量增加至57.73%, 木质素含量降低至28.29%, 表明Na2SO3预处理具有良好的脱木素能力。 此外, 经过碱性亚硫酸钠协同处理后, 发现碳水化合物含量提高到72.71%, 木质素含量降低到26.34%。 与亚硫酸钠协同处理相比, 碱性亚硫酸钠协同处理具有更好的脱木素能力, 同时降解了部分木聚糖, 大大增加了纤维素在生物质表面的暴露面积, 有助于纤维素酶对纤维素的生产性吸附。 这表明添加NaOH有助于除去其他成分(即木质素, 半纤维素和提取物等), 暴露并保留更多纤维素。 此结论与先前的研究吻合[8]。
2.2 泡桐样品的X射线衍射(XRD)分析
为研究预处理过程中样品的物理结构变化, 通过XRD测定处理前后泡桐样品的纤维素结晶度。 图1为预处理前后泡桐样品的X射线衍射图谱, 图中101, 002及040峰是典型的纤维素I的特征, 其衍射角分别为16.02°, 22°和34.76°。 经过预处理后, 002峰位置均向右有所偏移, 说明经过预处理后结晶区晶胞参数变小, 晶面间距变小, 同时发现, 碱性亚硫酸钠协同处理后, 002峰尖锐程度最大, 衍射峰明显增强, 纤维素结晶度CI明显高于其他样品。

图1 预处理前后泡桐样品的X射线衍射图
纤维素结晶度指数是由X射线衍射仪测得的光谱峰高计算所得[10]。 如表1所示, 未处理泡桐的结晶度是40.23%。 这可能归因于部分无定型物质的存在, 如木质素和半纤维素等。 经过乙酸处理后, 泡桐样品的结晶度增加至47.25%。 这可能是由于乙酸降解掉了一部分木聚糖, 从而导致纤维素结晶度增加。
亚硫酸钠协同处理后, 样品结晶度增加至50.32%。 预处理固体纤维素结晶度的增加可能归因于部分木质素, 木聚糖和其他组分的降解脱除, 从而降低整体无定形特征。 碱性亚硫酸钠协同处理导致更高的结晶度出现, 这可能是由于氢氧化钠的引入促进了更多木质素和其他成分被去除。 预处理后样品纤维素结晶度的增加, 表明更多无定型组分被脱除, 这与组分中葡聚糖含量的增加规律一致。
2.3 红外光谱分析
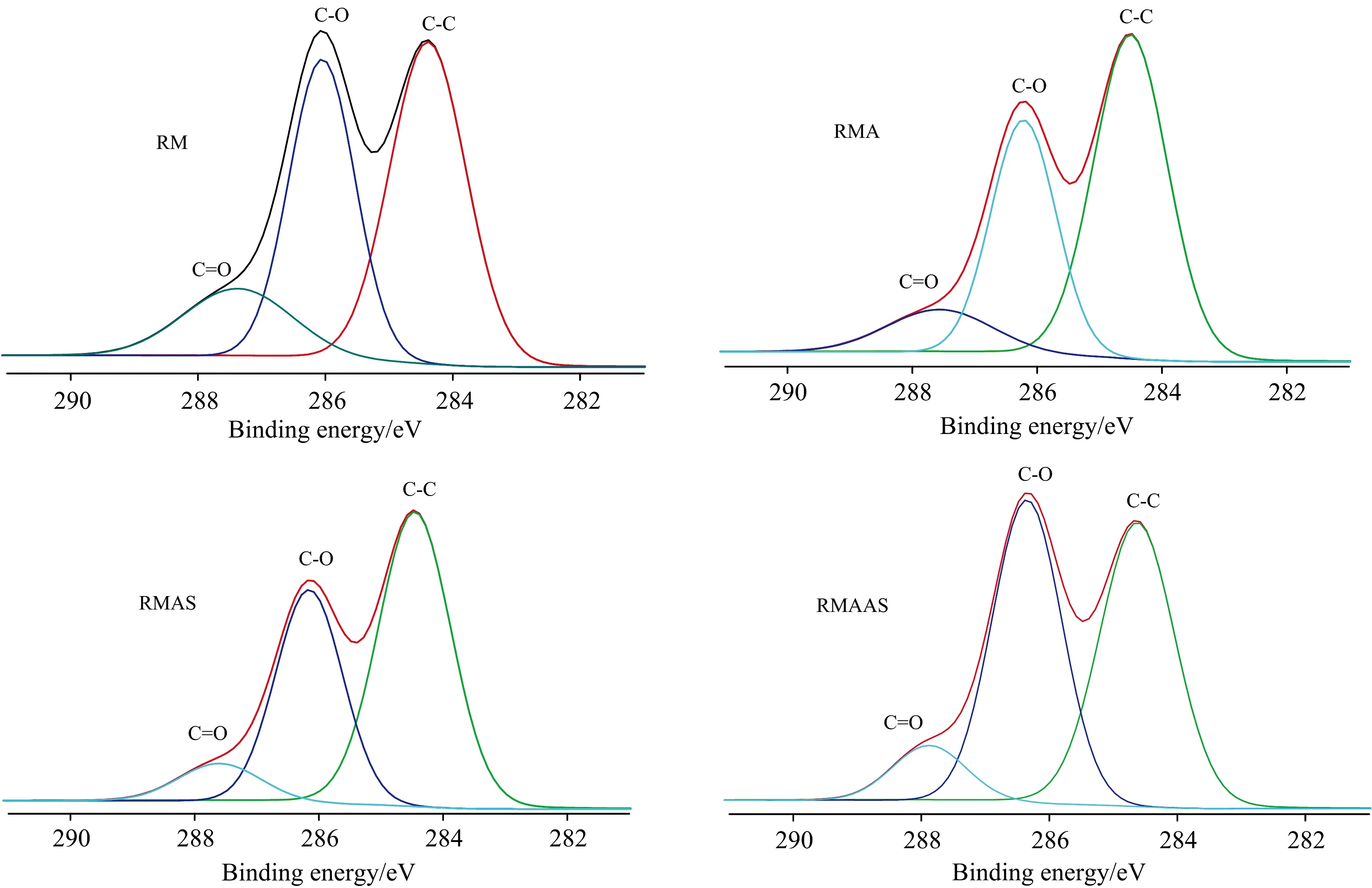
预处理前后泡桐的红外光谱图谱如图2所示。 1 731 cm-1处的吸收峰代表半纤维素的乙酰基和糖醛酯基或木质素中阿魏酸与香豆酸中羧基的酯键连接[10]。 在1 731 cm-1处, 乙酸处理后泡桐的吸收强度增加, 可能是由于乙酸脱除了部分木聚糖组分导致木质素含量增加。 两次协同处理后吸收接近消失, 吸收强度的顺序是RMA>RM>RMAS>RMAAS。 这表明协同处理后木质素可能发生了降解。 木质素特征吸收越少表明其结构破坏越大, 从而使得纤维素酶更容易接近纤维素[11]。 化学结构的破坏导致泡桐样品的顽抗性降低, 后续酶促水解性增强[8]。 碱性亚硫酸钠协同处理表现出更强的破坏底物结构的能力。 此外, 纤维素的特征吸收峰2 900, 3 338及1 059 cm-1, 协同处理后泡桐样品的吸收强度明显增加。 同时观察到, 木质素的特征峰在1 249 cm-1(C—O愈创木基环)和1 636 cm-1处, 乙酸处理样品的木质素峰值强度明显高于协同处理后样品的峰值强度。 这可能是由于协同处理使得泡桐原料的木质素结构进一步被破坏。 相比较于未处理的泡桐原料, 预处理后的泡桐在898和1 159 cm-1两处峰值强度(β-D糖苷键的特征峰)均有所提高, 峰强度的顺序是RM 图2 预处理前后泡桐样品的傅里叶红外光谱 图3为所有泡桐样品的X射线光电子能谱图谱。 可以发现, 经过碱性亚硫酸钠协同处理后, C—C含量明显减少, C—O含量明显增加。 这表明碱性亚硫酸钠协同处理对于样品表面木质素的脱除十分有利。 所有泡桐样品的氧碳比(O/C)是根据生物质材料表层(2~10 nm)的碳氧组成计算的(表2)。 据报道, 不同生物质组分的O/C比值按以下顺序排列: 纤维素(约为0.83)>半纤维素>木质素>抽提物(即脂肪酸, 烃类等)。 木质素和抽提物的理论O/C比分别为0.33和0.09[12]。 未处理泡桐的O/C比为0.45。 乙酸处理后, 泡桐样品的O/C比下降为0.387, 较为接近木质素的理论值0.33。 这可能是由于乙酸处理过程中降解了部分木聚糖, 表面木质素含量相对增加导致。 经亚硫酸钠协同处理后, O/C比增加到0.39, 仍然非常接近木质素的理论值(0.33)。 一般情况下, 预处理过程可以去除大量的抽提物。 因此, 数据表明亚硫酸钠协同处理泡桐的表面仍有大量木质素, 尽管木质素总量明显减少(表1), 可能是由于Na2SO3预处理和洗涤过程中木质素的再沉积。 高的O/C表示样品表面纤维素和(或)半纤维素含量较高, 低的O/C表示样品表面木质素较多[13]。 因此, O/C的增加表明表面木质素含量减少。 经碱性亚硫酸钠协同处理后, O/C提高到0.46, 说明表面木质素含量明显减少。 因此, 碱性亚硫酸钠协同处理更有利于总木质素的脱除及表面木质素的去除。 正如组分分析部分所讨论的, 碱性亚硫酸钠协同处理后泡桐的碳水化合物含量明显增加, 更加有助于酶水解。 图3 预处理前后泡桐样品的X射线光电子能谱图谱 (1)碱性亚硫酸钠协同处理表现出最佳的预处理效果。 葡聚糖相对含量增加至67.48%, 木质素及木聚糖等酶水解屏障性组分均有很大比例降解脱除。 表2 预处理前后泡桐样品的光电子能谱分析结果 Table 2 The O/C ratio and concentration of functionality by percentage carbon inPaulowniasamples before and after pretreatment 预处理方法O/CC1/%C2/%C3/%RM0.45048.09137.87514.034RMA0.38756.00034.5009.500RMAS0.39055.40036.3008.300RMAAS0.46046.70044.8008.500 (2)表征分析结果与组分分析规律一致。 FTIR分析显示, 碱性亚硫酸钠协同处理对样品结构破坏最大, 木质素特征吸收减弱, 纤维素特征吸收增强。 XPS分析可见, 碱性亚硫酸钠协同处理可以移除更多的表面木质素, 暴露并保留更多的表面碳水化合物(O/C比增加, C1含量减少, C2含量增加)。 XRD分析表明, 预处理后样品的纤维素结晶度均有不同程度增加, 002峰右移。 碱性亚硫酸钠协同处理后, 衍射峰衍射强度明显增强, 峰形变得高且尖锐, 更多无定型组分(木质素及半纤维素等)被脱除, 从而有助于提高后续的纤维素酶解效率。
2.4 X射线光电子能谱(XPS)分析

3 结 论

