改性黄原胶酯化条件的优化及结构的表征
2020-02-25吴君波江容安
王 珂, 杨 波*, 杨 光, 吴君波, 江容安
1.上海理工大学医疗器械与食品学院, 上海 200093;2.上海市杨浦区中心医院, 上海 200090
黄原胶是经黄单胞杆菌发酵产生的微生物阴离子多糖,由D-葡萄糖、D-甘露糖、D-葡萄糖醛酸、乙酰基和丙酮酸构成[1],相对分子质量在2×106~5×107之间[2]。黄原胶在分子间和分子内可通过氢键等作用力形成螺旋和多重螺旋刚性结构[3-4],具有增稠、耐温、耐酸碱等稳定性质[5]。但黄原胶在溶解过程中容易导致外层迅速吸水膨胀,阻止水分与内层接触,从而出现结团现象,溶解性较差[6]。
研究表明,通过一些物理、化学改性可以改变黄原胶的分子结构,提高黄原胶的溶解速度。黄原胶在物理改性方法中,超声波降解[7]、辐射降解[8]、机械降解[9]等可以使黄原胶分子链断开,但粘度降低。黄原胶在化学改性方法中,YAHOUM等[10]将黄原胶与一氯乙酸进行醚化反应,得到溶解性较好的改性产物,但粘度也相应降低。SU等[11]通过化学改性将甲醛取代黄原胶分子链上的羟基,使分子间的作用力和结构发生变化,速溶性和粘度得到提高。HAMCERENCU等人[12]将黄原胶分别与丙烯酸和丙烯酰氯、马来酸酐进行反应,酯化反应的效果更好。大多数化学改性黄原胶似乎都是应用于采油等工业方面,从提高速溶性和粘度的目的上对食品级黄原胶进行化学改性且对其毒理学方面的研究较少。
本研究主要通过响应面优化食品级黄原胶与马来酸酐酯化的反应条件,且该改性方法操作简单方便;然后对改性黄原胶进行结构表征,分析粘度和速溶性提高的原因。最后以酵母为对象进行初步的细胞毒性实验[13],为进一步探究是否可应用于食品、医药等行业做铺垫。
1 材料与方法
1.1 材料
1.1.1实验材料
黄原胶(食品级,含水量11%),山东阜丰发酵有限公司;马来酸酐(又名顺丁烯二酸酐),上海沃凯生物科技有限公司;四氢呋喃(THF)、95%乙醇,国药集团化学试剂有限公司;安琪酵母,安琪酵母股份有限公司;葡萄糖、蛋白胨,国药集团化学试剂有限公司;四氮唑盐(MTT),北京博奥拓达科技有限公司;二甲基亚砜(DMSO),北京索莱宝科技有限公司。
1.1.2实验仪器
NDJ-5S粘度计,上海昌吉地质仪器有限公司;LC-MSH-PRO磁力搅拌器,邦西仪器科技有限公司;DZF-6030A真空干燥箱,上海一恒科学仪器有限公司;Nicolet is10型傅里叶红外光谱仪,美国产;Agilent 1260型高效液相色谱仪,DAWN HELEOS-II型多角度激光光散射仪,Optilab T-rEX示差检测器,美国Wyatt公司;XRD-6100型X-射线衍射仪,日本岛津;Read Max 1900型光吸收全波长酶标仪,上海闪谱生物科技有限公司。
1.2 实验方法
1.2.1黄原胶与马来酸酐的酯化反应
将一定量的马来酸酐溶解于40 mL四氢呋喃中,加入3.2 g黄原胶,室温下溶胀1.5 h,水浴加热并冷凝回流一段时间;反应结束后,过滤去除四氢呋喃,剩余物倒入15 mL 90%乙醇,醇沉、离心,弃去上清液,重复三次,测pH;40 ℃真空干燥3 h,确定水分含量,约11%。
以黄原胶与马来酸酐的摩尔比、反应温度和反应时间为单因素,以取代度(Degree of Substitution,DS)为优化指标,采用响应面方法对改性工艺进行优化。
1.2.2改性黄原胶的速溶性、粘度的测定
取0.2 g改性黄原胶粉末慢慢加入正在搅拌的100 mL蒸馏水中,800 r/min磁力搅拌至完全溶解,记录溶解时间。将完全溶解的0.2%改性黄原胶溶液用旋转粘度计测定粘度,单位mPa·s。
1.2.3改性黄原胶的取代度的测定
测定酯化反应的DS根据文献[14]的方法并稍作改动。称取0.2 g改性黄原胶和10 mL 75%乙醇于锥形瓶中,55 ℃水浴加热30 min,加入30 mL 0.1 mol/L的NaOH标准溶液,55 ℃水浴加热15 min。加2滴1%酚酞指示剂,用0.1 mol/L的HCl标准溶液滴定,直至紫红色消失且不变色。以黄原胶为空白对照。
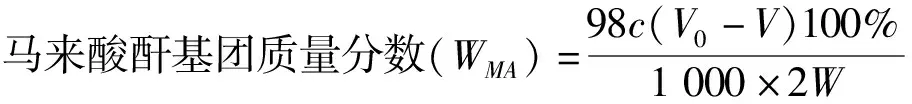
(1)
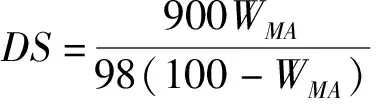
(2)
式(1)(2)中V0,空白组消耗HCl的体积,mL;V,实验组消耗HCl的体积,mL;c,HCl溶液的浓度,mol/L;W,样品的质量,g。
1.3 改性黄原胶的结构表征
1.3.1傅里叶红外光谱
将改性黄原胶粉末用溴化钾压片制样,使用美国产nicolet is10型傅里叶红外光谱仪测定,扫描波数(4 000~400)cm-1。
1.3.2尺寸排阻色谱结合多角度激光光散射
色谱柱:Shodex OHpak SB-805HQ型。测定条件:流动相0.1 mol/L NaCl溶液;柱温40 ℃;流速0.6 mL/min。用流动相配制0.2 mg/mL浓度的样品溶液,搅拌溶解,过0.22 μm滤膜后进样200 μL。
1.3.3X-射线衍射
样品粉末进行压片,采用XRD-6100型日本岛津的X-射线衍射仪进行测定。测定条件:Cu靶,管压40 kV,管流30 mA,扫描步长0.02°,扫描角速度6°/min,2θ角范围10~80。
1.4 细胞毒性实验
采用MTT法[15]检测改性黄原胶对酵母细胞活力的影响。取对数生长期酵母细胞悬液稀释,取100 μL于96孔板,加入100 μL改性黄原胶溶液,使其浓度分别为0.025%、0.050%、0.100%、0.200%和0.400%,对照组加无菌水。于5%CO2、37 ℃培养36 h,加入10 μL MTT溶液,继续培养4 h后加100 μL DMSO,在OD490处检测吸光度。MXG溶液对酵母细胞活力若有提高,可用增长率表示,如公式(3):
酵母细胞增长率(%)=
(3)
2 结果与分析
2.1 黄原胶与马来酸酐的酯化改性结果
2.1.1黄原胶与马来酸酐摩尔比对酯化反应结果的影响
黄原胶(xanthan gum, XG)与马来酸酐(maleic anhydride, MA)摩尔比为1∶1、1∶6、1∶11、1∶16和1∶21,将黄原胶的量固定为3.2 g,反应温度和时间设为66 ℃和24 h。
由图1可知,随着马来酸酐的量增加DS也增大,在MXG∶MMA=1∶11时达到最大,为0.37,随后趋于稳定。而粘度也与之相似,0.2%的改性黄原胶溶液在MXG∶MMA=1∶11时达到630 mPa·s,溶解时间为3 min~4 min;而未改性的黄原胶溶液溶解时间12 min,粘度为220 mPa·s,即改性黄原胶比黄原胶在速溶性和粘度上均提高了近3倍。说明黄原胶与马来酸酐摩尔比在1∶11时达到较好的效果,随着马来酸酐的量增加,速溶性和粘度都处于相对稳定的状态。与理论上11个马来酸酐与1个黄原胶单元结构中的11个羟基反应基本一致。
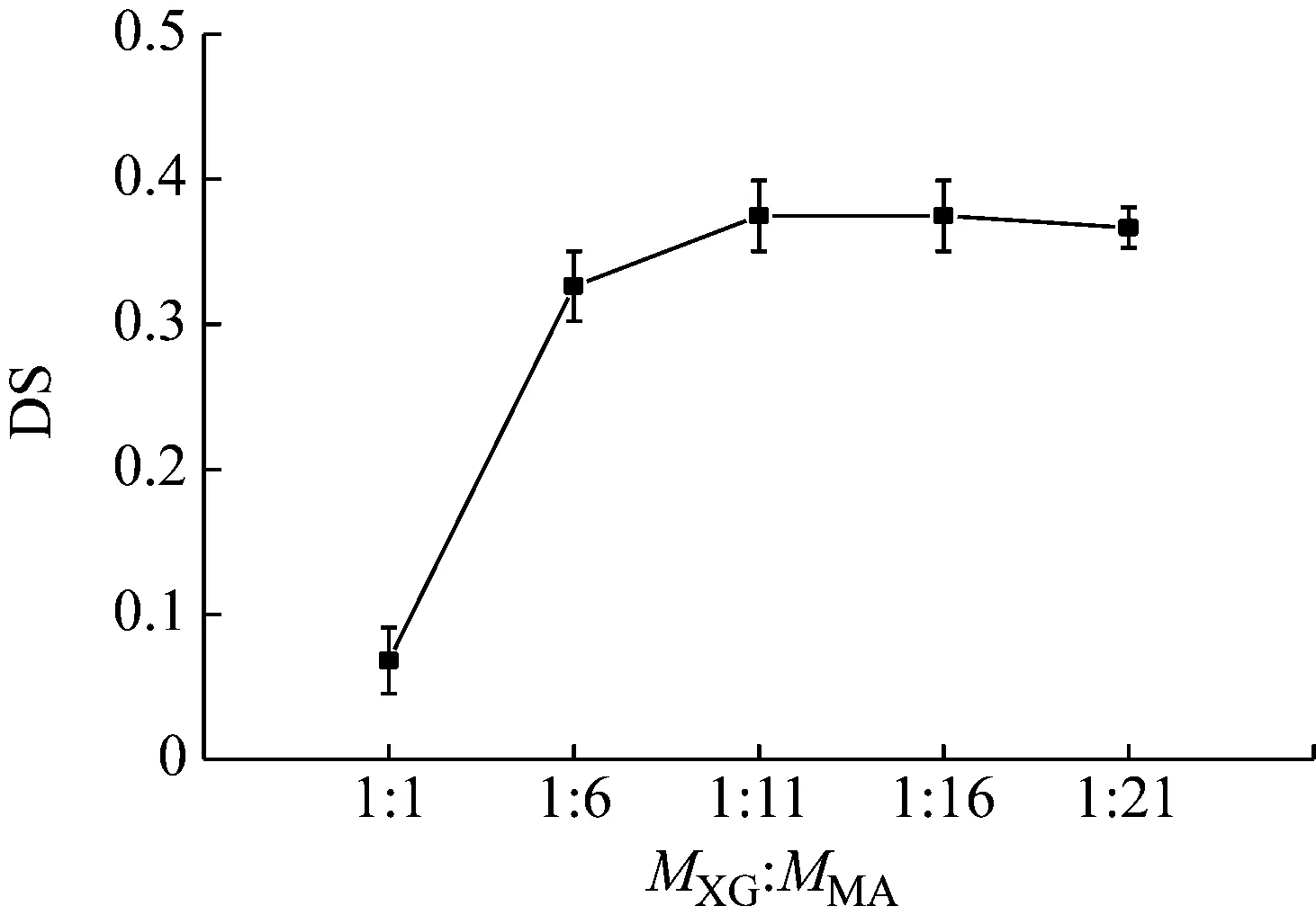
图1 黄原胶与马来酸酐摩尔比对DS的影响
2.1.2反应温度对酯化反应结果的影响
由于反应溶剂四氢呋喃的沸点为66 ℃,反应温度分别设为26 ℃、36 ℃、46 ℃、56 ℃和66 ℃,黄原胶与马来酸酐摩尔比为1∶11,反应时间为24 h。
根据图2,在26 ℃至56 ℃之间,DS并无明显变化,而上升至66 ℃时,DS明显增加。结合速溶性和粘度的角度来看,26 ℃至56 ℃变化不大,0.2%的改性黄原胶在7 min~10 min溶解,粘度在(240~290)mPa·s之间;而反应温度为66 ℃的改性黄原胶的溶解时间为3 min~4 min,粘度达到630 mPa·s。
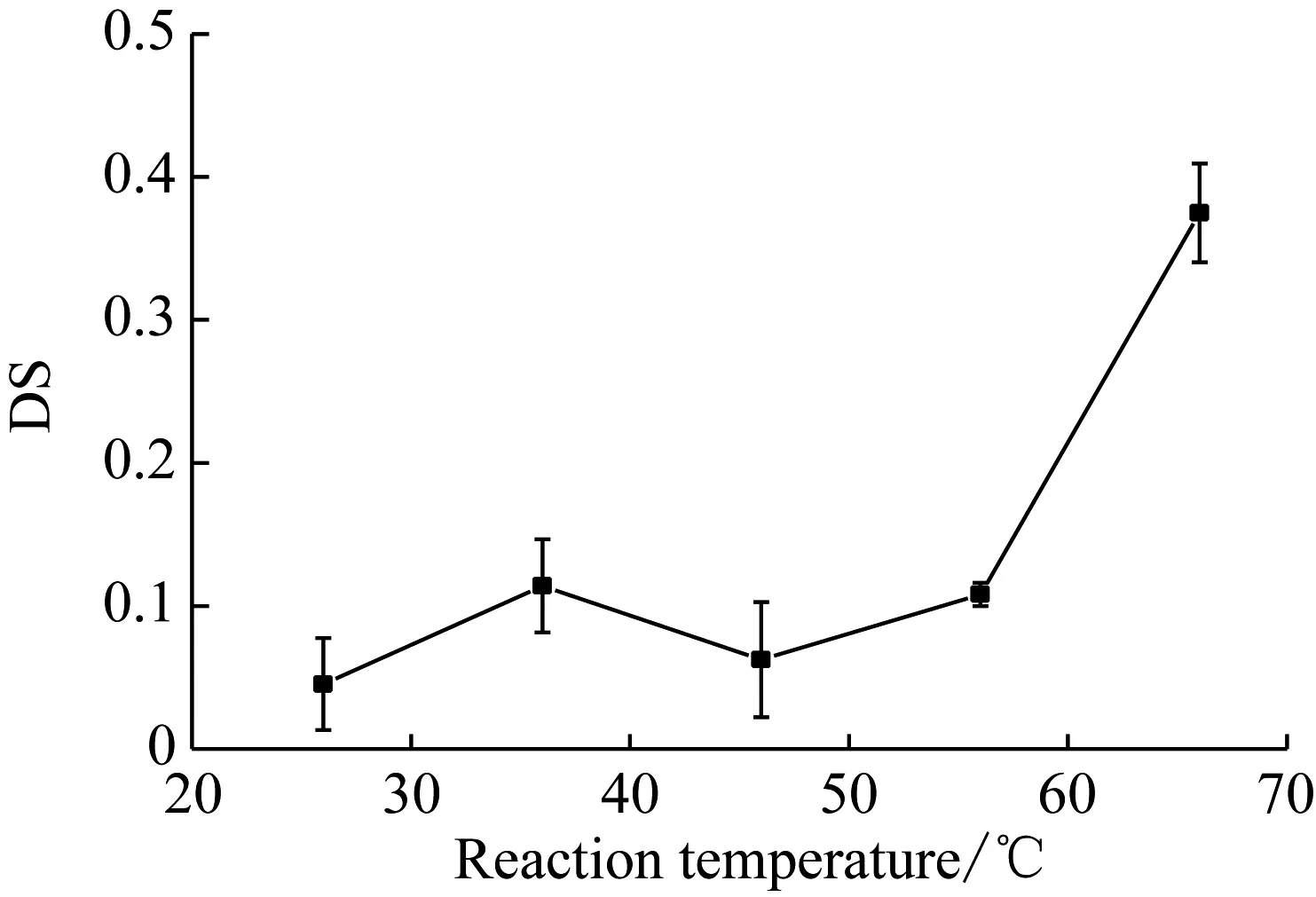
图2 反应温度对DS的影响
2.1.3反应时间对酯化反应结果的影响
反应时间分别设为6 h、12 h、18 h、24 h和30 h,黄原胶与马来酸酐摩尔比为1∶11,反应温度为66 ℃。
观察图3,反应时间从6 h至24 h时,改性黄原胶的DS逐渐上升,而30 h时略微下降;粘度的变化趋势与DS相似,推测30 h DS和粘度下降的原因可能是反应过程中伴随黄原胶的降解反应。王世高[16]在进行黄原胶的改性时讨论过,在80 ℃时,较多的黄原胶发生分子链断裂,导致粘度急剧下降。虽然本研究的温度为66 ℃,但长时间的加热也可能发生降解。
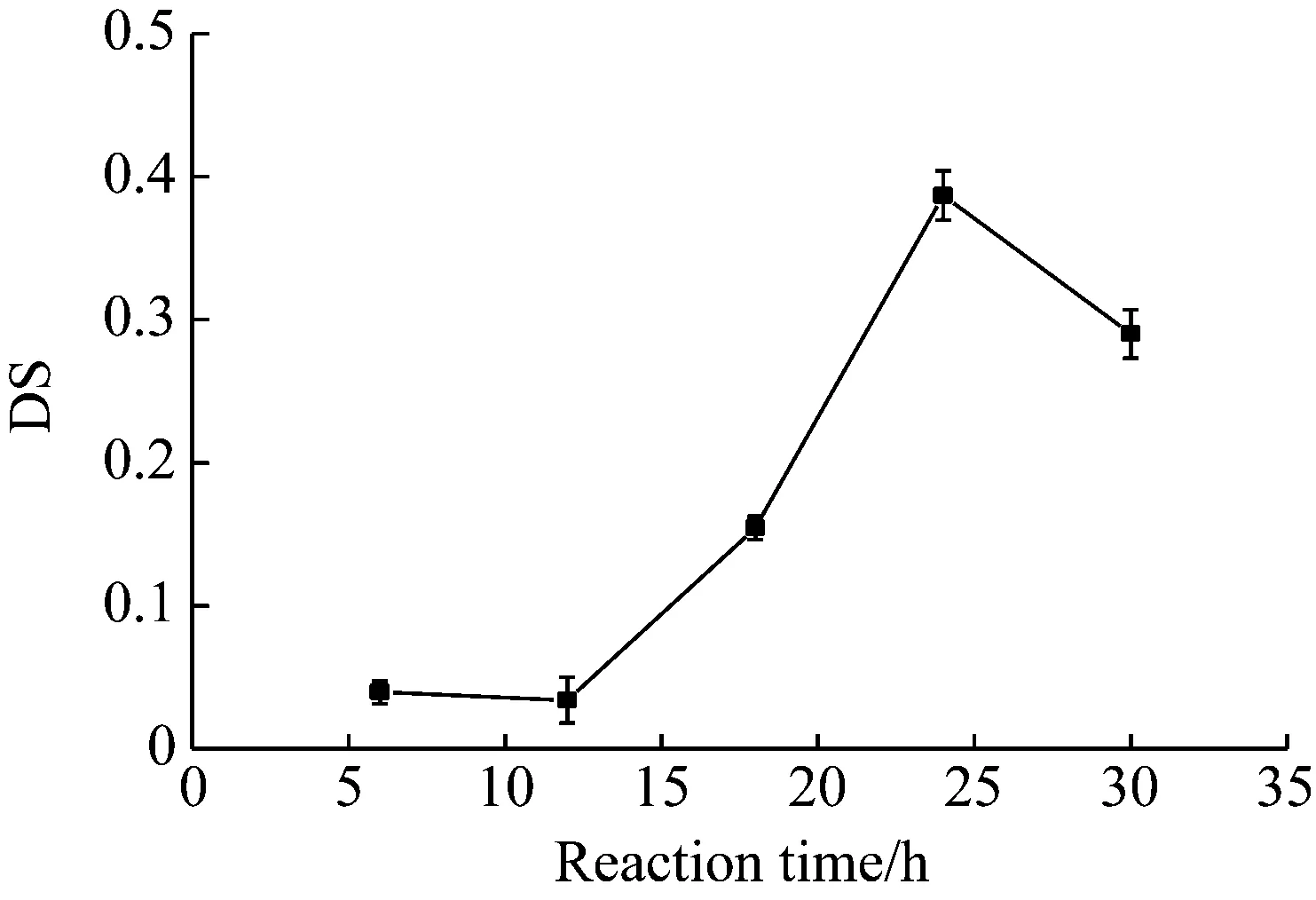
图3 反应时间对DS的影响
2.1.4响应面优化
因实验中反应温度不能超过溶剂的沸点,最终以66 ℃为最适反应温度,黄原胶与马来酸酐酯化反应的响应面设计如表1和表2。
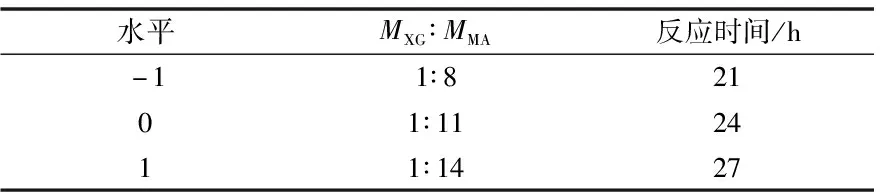
表1 黄原胶与马来酸酐酯化响应面设计因素和水平
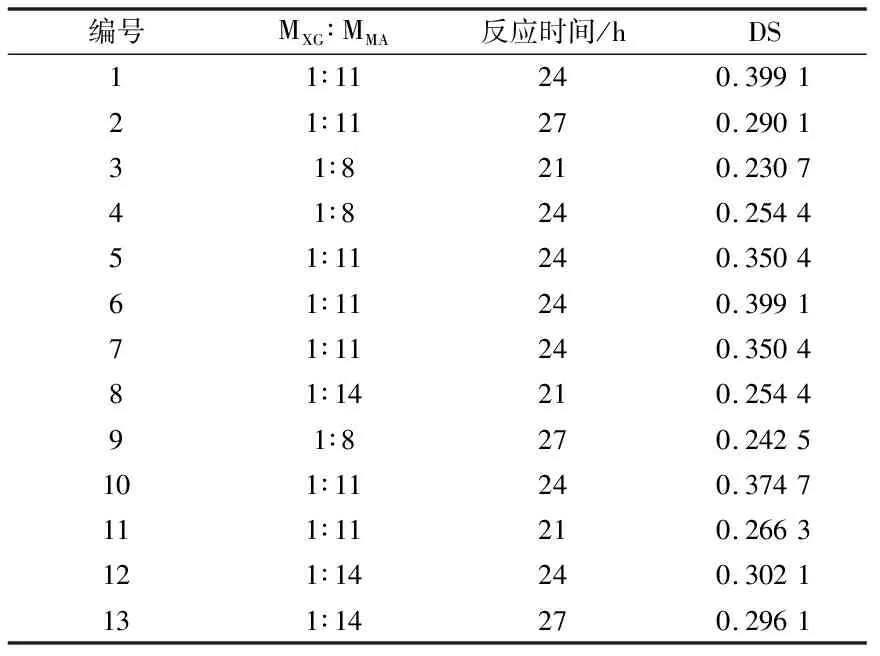
表2 黄原胶与马来酸酐酯化响应面设计实验结果
由表3可知,黄原胶与马来酸酐摩尔比(A)与反应时间(B)对酯化反应的影响程度A>B,试验模型的P<0.05,有显著性差异,失拟项为0.236 4,说明实验与实际情况拟合度较好。二次回归拟合方程:DS=0.36+0.021×A+0.013×B+7.475E-003×A×B-0.061×A2-0.061×B2,得到黄原胶与马来酸酐的最佳反应条件为马来酸酐的量为3.86 g,即MXG∶MMA=1∶11.5,反应时间为24.4 h,与理论基本相符合。
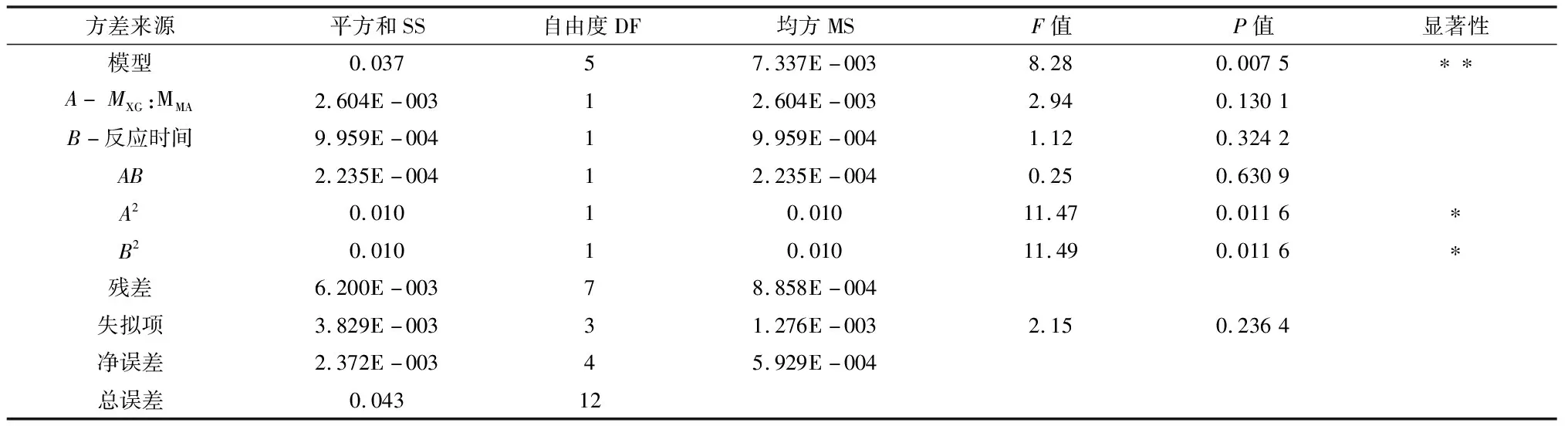
表3 回归模型的方差分析
注: *差异显著(P<0.05);**差异极显著(P<0.01)
验证最优条件下改性黄原胶(modified xanthan gum,MXG)的粘度达到了650 mPa·s,最后将黄原胶与改性黄原胶的一些性质进行对比,如表4。比较杜冠乐等人[17]研究的改性黄原胶,在0.2%浓度时,改性黄原胶的粘度比原料增加不到两倍,而本研究在粘度上增加了近3倍。

表4 黄原胶与改性黄原胶的性质对比
2.2 改性黄原胶的结构表征
2.2.1红外光谱
图4为黄原胶与改性黄原胶的红外光谱,改性黄原胶的吸收峰与黄原胶比较吻合,说明改性黄原胶保留了黄原胶骨架的基本结构。由图可知,在3 452 cm-1处为MXG的—OH伸缩振动吸收峰,相对XG(3 410 cm-1)发生42 cm-1的蓝移现象,说明改性黄原胶的分子间或分子内氢键作用力减小[18];2 922 cm-1为MX的—CH2的伸缩振动,MXG的—CH2伸缩振动吸收峰出现在2 925 cm-1。黄原胶分子上的羟基与马来酸酐反应形成酯键,同时马来酸酐开环形成羧基,MXG在1 266 cm-1处—OH的弯曲振动吸收峰明显减弱,主要原因是发生了酯化反应[19];在1 734 cm-1处为XG的酯羰基的伸缩振动,而MXG出现在1 735 cm-1处,推测有新的酯键导致峰位移;MXG在1 654 cm-1处出现—C=C—的伸缩振动,在2 517 cm-1出现新的羧基的—OH伸缩振动吸收峰,说明酯化改性成功。

图4 XG与MXG的红外光谱图
2.2.2相对分子量
运用光散射测得的结果如表5所示,黄原胶的平均相对分子量Mw为 6.568×106,Mr分布(Mw/Mn)为1.266,改性后Mw为9.118×106,Mr分布为1.175。改性后黄原胶的Mw提高了38.8%,主要原因是马来酸酐开环取代黄原胶分子上的羟基,也有可能少量酯化后形成的羧基与黄原胶分子发生了交联反应,使分子量增大[20],改性黄原胶粘度增大。改性后的Mr分布降低,说明MXG具有较少的不同分子量片段[21],且MXG比XG的Rg/Rh(Rg表示均方回转半径,Rh表示流体力学半径)小,说明黄原胶的分子结构比较疏松,分布不均匀,改性后黄原胶的结构比较紧凑,分布更加均匀。

表5 XG及MXG的相对分子量测定结果
2.2.3X-射线衍射
从图5中可以看出XG和MXG的完全无定形特征,且衍射曲线基本相似,说明两者的基本结构相似。其中XG在31.46°和75.14°处出现结晶区特征峰,表明改性后的黄原胶晶体结构发生改变。通过MDI jade软件计算两者的结晶度,XG为26.17%,MXG为21.27%,XG的结晶度较大。在无定形区域,分子间的相互作用力较弱,溶解时更容易分散在溶剂中,而结晶区的分子结构较为紧密,溶解速度较慢[22]。因此从峰型和结晶度可以得出,MXG比XG的结构更均匀,没有结晶区,在溶剂中具有更好的分散和溶解能力。
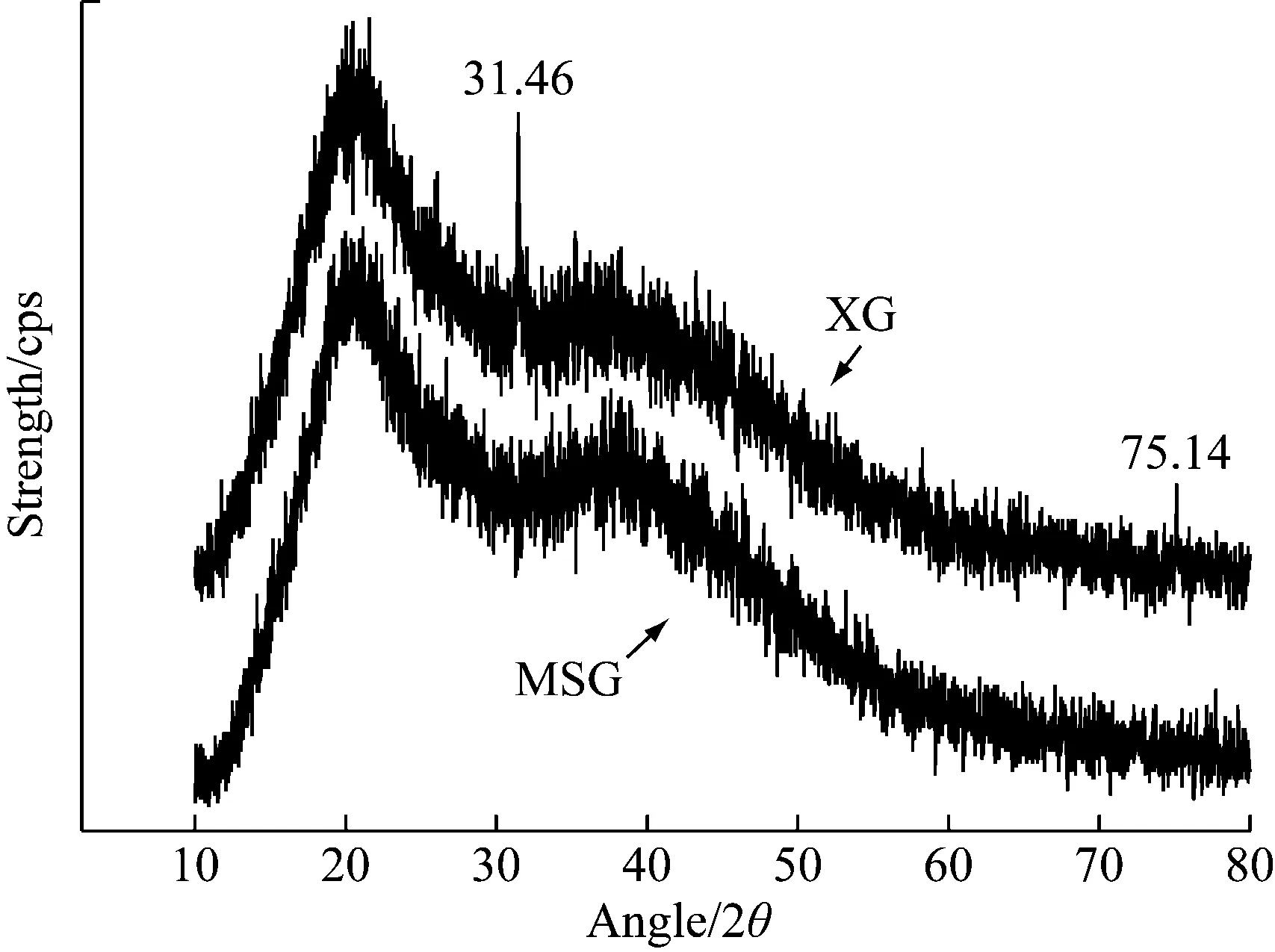
图5 XG与MXG的X-衍射图谱
2.3 细胞毒性实验结果分析
如图6所示,以不添加MXG溶液的酵母菌液为对照,添加MXG溶液的酵母细胞都有增长现象,且随着MXG溶液浓度升高,酵母细胞数量增多。高浓度MXG溶液对应增长率升高缓慢,原因可能是0.2%和0.4%浓度的溶液比较黏稠,对细胞的快速生长和繁殖会产生一定影响[23]。总的来说,MXG溶液不仅对酵母细胞没有抑制作用,还可以提高细胞活力,其主要原因为MXG属于微生物多糖,与XG相似,所以可以作为酵母菌生长所需的营养物质。
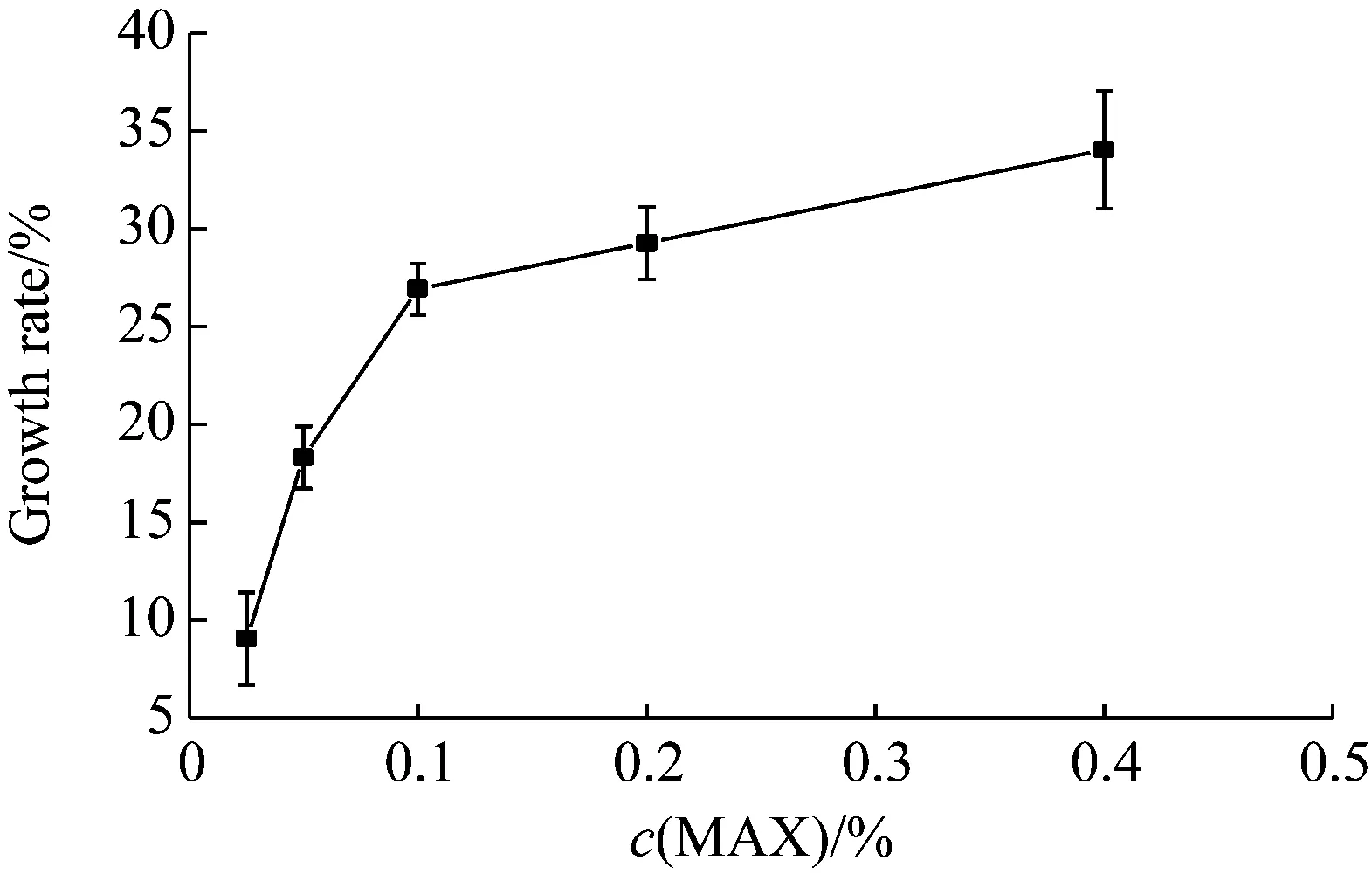
图6 不同浓度的MXG溶液对酵母细胞的影响
3 结论
通过单因素、响应面优化方法得到的黄原胶与马来酸酐酯化反应的最适方案,并进行结构表征分析和细胞毒性实验的结果如下:
(1)黄原胶与马来酸酐反应的最适条件为:MXG∶MMA=1∶11.5,反应时间24.4 h,反应温度66 ℃。0.2%改性黄原胶的速溶性和粘度提高近3倍。
(2)从红外光谱可以看出黄原胶改性后的—OH振动吸收峰从3 410 cm-1移至3 452 cm-1处,且在1 654 cm-1和2 517 cm-1出现新的特征吸收峰,说明酯化改性成功。结合相对分子量和X-射线衍射结果,改性后黄原胶的分子量增大,黄原胶分子内或分子间的氢键作用力和分子间相互作用力减小,减缓了溶解时分子外层迅速抱团的情况,从而加快了溶解速度。
(3)细胞毒性实验的结果表明改性黄原胶对酵母细胞没有毒性,并且作为多糖物质可以促进细胞生长。
