不同产地甘草中甘草苷等五种成分的含量比较研究
2020-01-18张黎娟汪盈盈
张黎娟,汪盈盈,李 飞
(1.亳州职业技术学院 药学院,安徽 亳州 236800 ;2.安徽中医药科学院 亳州中医药研究所,安徽 亳州 236800;3.亳州市食品药品检验中心,安徽 亳州 236800)
甘草来源于豆科(Leguminosae)植物甘草(Glycyrrhiza uralensisFisch.)、胀果甘草(G.inflataBat.)和光果甘草(G.glabra L.)的干燥根和根茎。具有补脾益气、祛痰止咳、缓急止痛、清热解毒、调和诸药的功效[1-2]。甘草是神农本草经记载的上品常用中药,在中国甘草应用于临床的时间已有4 000多年[3]。现代研究发现甘草的化学成分有三萜类、黄酮类、生物碱、多糖、氨基酸等。随着对甘草应用的不断深入研究,中药纳入医保使用范围,甘草的消耗量不断增加,导致甘草的市场需求量不断增加,并远超国内的野生产量,采取野生地区计划性人工种植后,仍无法满足国内外市场需求。除中国外,中东、中亚及东欧一些国家也拥有丰富的野生甘草资源,2008年起我国施行了进口甘草免关税,2011年我国甘草进口量为10 659 t,同比增长123 %,进口额为1 012万美元,同比增长160 %。进口甘草占到我国76 %的甘草进出口贸易量,已成为我国进口中药材饮片第二大品种[4]。
中药甘草的质量和影响质量的因素一直是备受关注的研究对象,目前闫永红、林寿全等[5-7]对国产野生与栽培甘草不同来源、不同产地、不同生长年限所导致的性状和活性成分含量的差异进行了研究,结果表明因来源、生态环境、生长年限等差异会造成甘草质量上的差别。国内外对甘草的研究多在于活性成分的药理作用和活性成分的提取分离[8-9]。因土壤、气候、来源等生长环境的差异,进口甘草与国产甘草质量是否一致,药用疗效是否相同,这都是进口甘草能否用于中医药临床必须解答的问题。对进口甘草在甘草苷、异甘草苷、甘草素、异甘草素、甘草酸5种活性成分上与国产甘草进行质量一致性评价,期望能发现其中的质量差异,为今后的进口甘草应用于中医临床和深入研究提供参考。
高晓娟等考证古代文献发现国产甘草的核心产区从以山西为主产区变迁为今天的以西北地区(内蒙古、甘肃、新疆、宁夏)为主要产区[10]。乌拉尔甘草分布于内蒙、甘肃、宁夏、新疆、青海、陕西、山西、河北、辽宁、吉林、黑龙江,胀果甘草分布于甘肃、新疆、陕西、山西,光果甘草分布于新疆、青海。林寿全等研究表明生态因子对甘草质量有影响,认为不同产区的甘草在性状和有效成分含量上有显著差异。在中东、中亚及东欧一些国家也拥有丰富的野生甘草资源,例如土库曼斯坦、乌兹别克斯坦、哈萨克斯坦、阿塞拜疆、巴基斯坦、塔吉克斯坦、阿富汗等地皆有野生甘草资源分布,并向中国出口[3]。
1 仪器与材料
1.1 仪器
安捷伦1260高效液相色谱仪(包括 G1311C四元单泵、G1329B 进样器、G1316A紫外检测器、Technoiogies工作站);十万分之一电子天平(上海仪科仪器有限公司);数显式电热恒温水浴锅(广州市海数实验仪器制造厂);700VDE型数控超声波清洗器(合肥金龙克机械制造有限公司);EV321真空旋转蒸发仪(LadTech);SHZ-DⅢ予华牌循环水真空泵(巩义市予华仪器有限责任公司);UORII-207优普系列超纯水器(四川优普超纯科技有限公司)。
1.2 材料
国产甘草购于亳州中药市场;进口甘草(乌兹别克斯坦、哈萨克斯坦、巴基斯坦、吉尔吉斯斯坦)来源于国家中药材质量监督检查中心,经蒋磊副主任中药师鉴定为胀果甘草(G.inflataBat);甘草苷对照品(批号18030905)、甘草酸铵对照品(批号18012902)、甘草素对照品(批号17112302)、异甘草苷对照品(批号17042102)、异甘草素对照品(批号17072104,成都普菲德生物科技有限公司);磷酸(批号151005,上海展云化工有限公司);甲醇(批号20170712)、无水乙醇(批号20170612)、乙腈(批号20180103),来源于国药集团化学试剂有限公司;实验用水均为纯化水。
2 方法与结果
2.1 色谱条件
色谱柱:岛津 Inerisil ODS-3(250 mm×4.6 mm,5 µm),0.85 %磷酸水溶液为流动相A,乙腈为流动相 B,梯度洗脱(见表1),柱温 25 ℃,检测波长237 nm,流速 1.0 mL/min[11]。上述色谱条件下,甘草苷、甘草酸、异甘草苷、甘草素、异甘草素对照品高效液相色谱图,见图1。
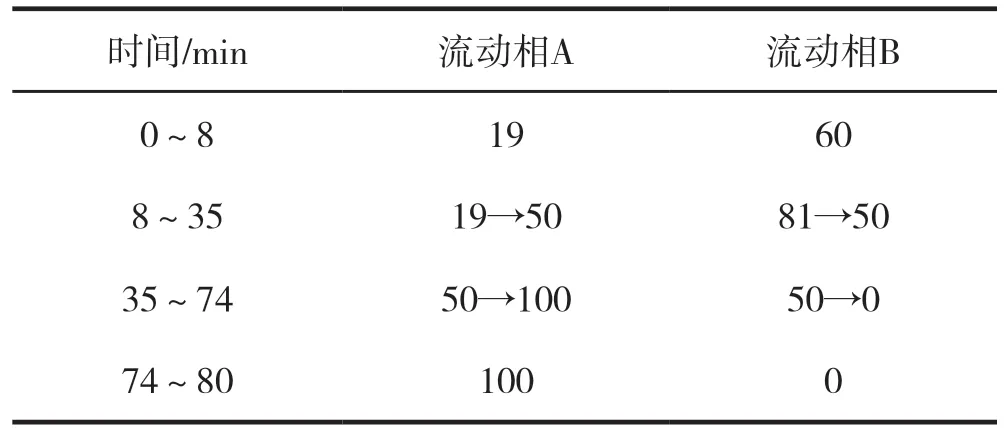
表1 梯度洗脱程序/%

图1 甘草混合对照品溶液HPLC图
2.2 供试品溶液的制备
取本品粉末(过三号筛)约2.0 g,精密称定,置 250 mL锥形瓶中,加入80%乙醇40 mL,浸泡提取3次,合并收集3次滤液,减压旋转蒸发至5 mL,加70 %乙醇定容于25 mL容量瓶中,即得。
2.3 对照品溶液的制备
分别精密称取减压干燥4 h的甘草苷对照品10.23 mg、异甘草苷对照品13.25 mg、甘草素对照品11.20 mg、甘草酸铵对照品11.21 mg、异甘草素对照品10.61 mg,置于50 mL容量瓶,用70 %乙醇定容至刻度线,分别精密吸取 2.0、2.0、0.5、2.0、1.0 mL,置 10 mL 容量瓶中,用 70%乙醇定容,摇匀,得浓度分别为 0.040 9、0.053 0、0.011 2、0.044 8、0.021 2 mg/mL 的混合对照品溶液。
2.4 方法学考察
2.4.1 线性关系考察
分别精密吸取混合对照品溶液 0.5、1、5、10、15 µL 进样,记录色谱峰面积,以进样量为横坐标,色谱峰面积为纵坐标,计算回归方程。甘草苷:Y=40.879X+12.134,r=0.998 5,线性范围 0.020 5~0.613 05 µg;异甘草苷:Y=48.4X+0.453 09,r=1,线性范围 0.026 5~0.705 0 µg;甘草素:Y=25.475X+2.000 05,r=0.999 9 线性范围 0.005 6 ~ 0.168 0 µg;甘草酸:Y=13.526X+1.299 09,r=0.999 07;线性范围 0.022 4~ 0.672 0 µg;异甘草素:Y=27.706X+0.735 08,r=1;线性范围 0.010 6~ 0.318 00 µg。各成分在线性范围内线性关系良好。
2.4.2 精密度试验
精密吸取同一批国产甘草供试品溶液 10 µL,连续进样 5 次,记录色谱峰面积。结果甘草苷、异甘草苷、甘草素、甘草酸、异甘草素峰面积 RSD 分别为1.74%、0.96%、1.27%、1.54%、1.69%。
2.4.3 稳定性试验
取国产甘草供试品溶液,分别在 0、2、4、8、16、24 h 进样 10 µL,记录色谱峰面积,RSD 分别为1.67 %、0.98 %、1.29 %、1.49 %、1.57 %,表明供试品溶液在24 h 内基本稳定。
2.4.4 重复性试验
取同一批国产甘草粉末5份,2.001 9 g、2.001 2 g、2.001 0 g、2.002 1 g、2.002 2 g、精密称定,制备5份试品溶液,进样 10 µL,记录色谱峰面积。甘草苷、异甘草苷、甘草素、甘草酸、异甘草素平均含量分别为 7.295、0.956、0.180、17.347、0.175 mg/g。RSD 分别为1.49 %、0.95 %、1.24 %、1.43 %、1.27 %。
2.4.5 加样回收率
取已知含量的同一批国产甘草粉末5份,2.002 5 g、2.001 4 g、2.001 5 g、2.000 3 g、2.002 0 g,精密称定,制备供试品溶液,每份加入相应质量的对照品,进样 10 µL分析。计算甘草苷、异甘草苷、甘草素、甘草酸及异甘草素平均加样回收率分别为98.98 %、99.45 %、96.97 %、99.46 %、96.85 %(见表2),实验结果表明本法回收率良好。
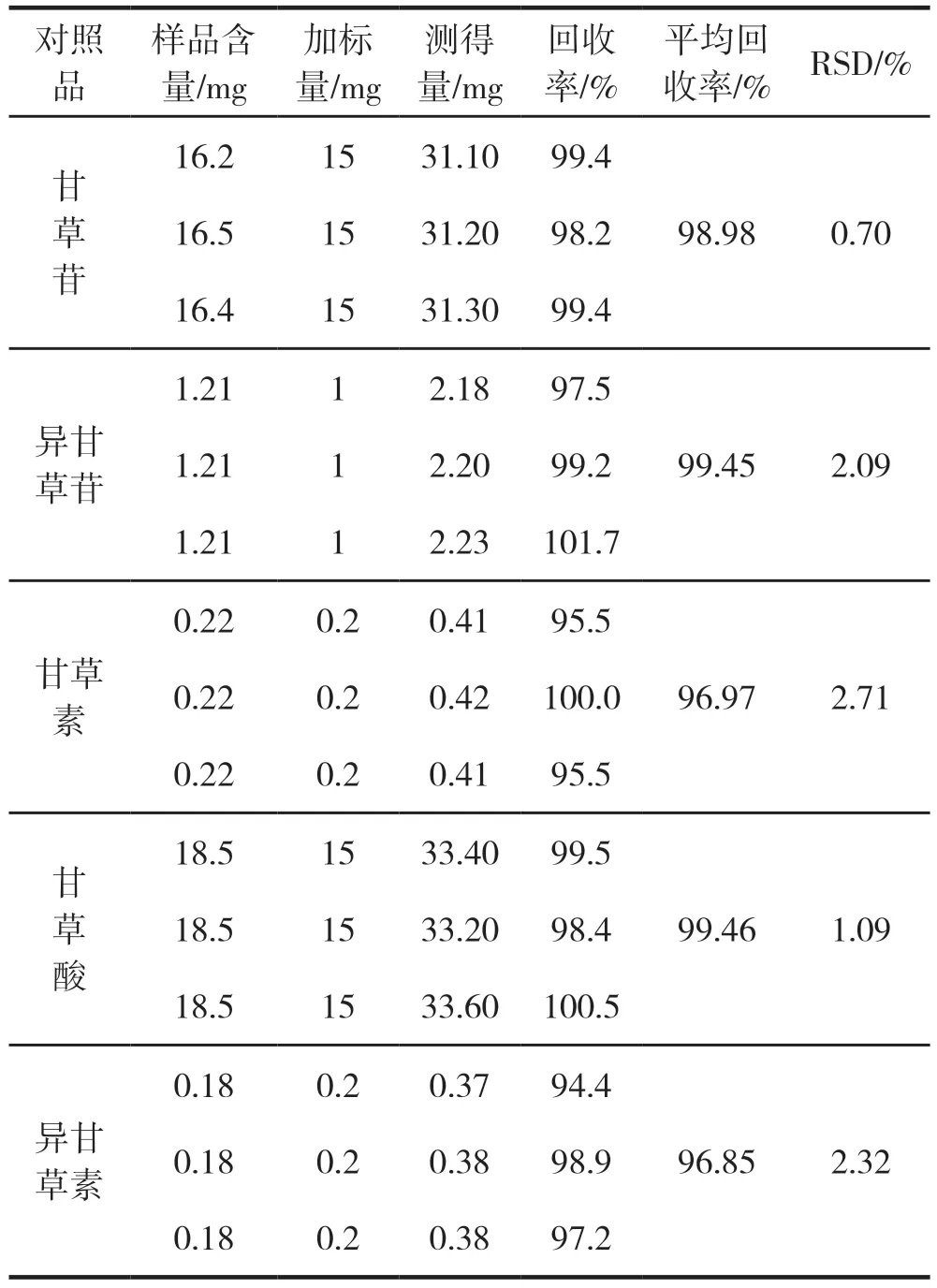
表2 加样回收率试验结果
2.5 甘草含量测定
取甘草苷、异甘草苷、甘草素、甘草酸、异甘草素混合对照品溶液,制备巴基斯坦进口甘草、哈萨克斯坦进口甘草、吉尔吉斯斯坦进口甘草、乌兹别克斯坦进口甘草、国产甘草供试品溶液,进样10 µL 分析。
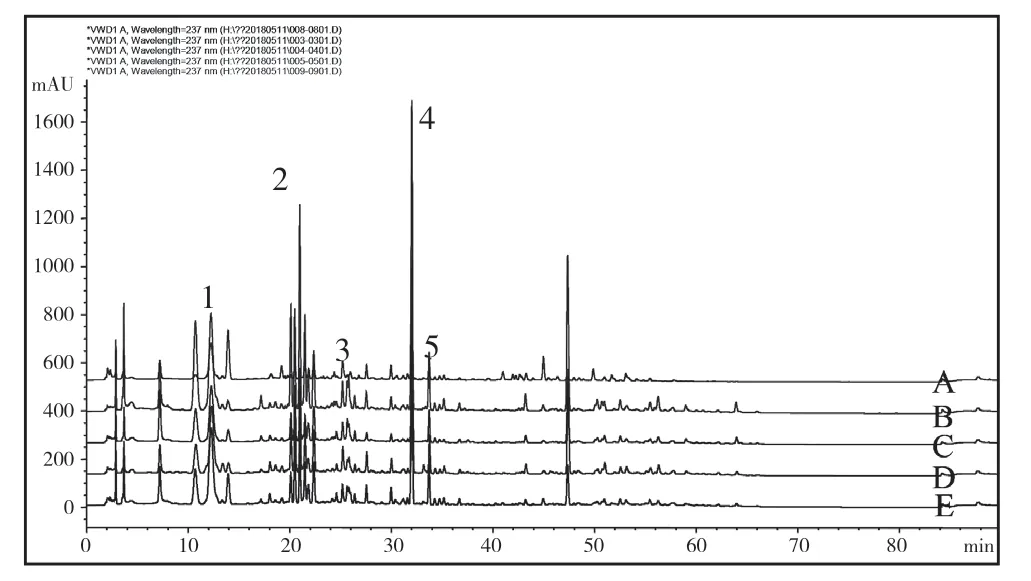
图2 不同产地的甘草色谱图色谱图
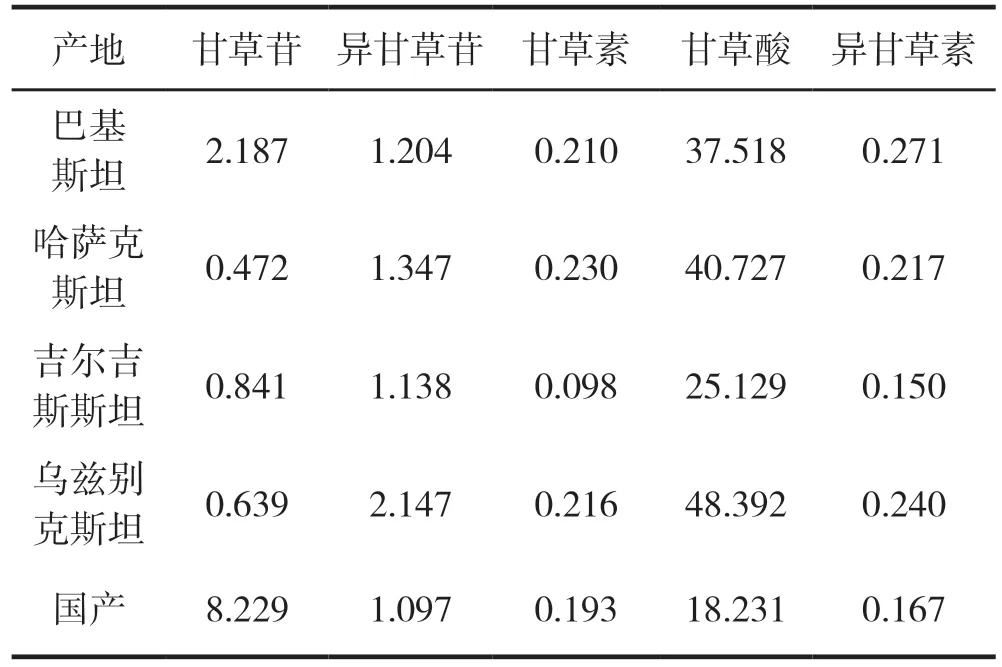
表3 进口甘草与国产甘草甘草苷、异甘草苷、甘草素、甘草酸、异甘草素含量测定结果/(mg/g)
3 讨论
实验对不同提取方法进行比较,温浸法提取甘草素、异甘草素、异甘草苷效果优于加热回流法和超声法,且甘草苷、甘草酸的提取效果亦良好。确定了色谱柱为 ODS-3(250 mm×4.6 mm,5 µm),0.85 %磷酸水溶液和乙腈为流动相,梯度洗脱,柱温25 ℃,检测波长237 nm,流速1.0 mL/min的色谱条件测定甘草素、甘草苷、异甘草素、甘草酸、异甘草苷5种活性成分结果准确,可靠。
本实验测定了进口甘草和国产甘草中的甘草苷、异甘草苷、甘草素、甘草酸、异甘草素的含量。测定结果表明甘草苷含量以国产甘草最高。进口甘草与国产甘草在甘草素成分上存在明显质量差异。
