黑龙江省稻瘟病菌无毒基因AVR-Pib、AVR-Pik和AvrPiz-t的检测与分析
2020-01-14孟峰张亚玲靳学慧张晓玉姜军
孟峰,张亚玲,靳学慧,张晓玉,姜军
黑龙江省稻瘟病菌无毒基因AVR-Pib、AVR-Pik和AvrPiz-t的检测与分析
孟峰,张亚玲,靳学慧,张晓玉,姜军
(黑龙江八一农垦大学农学院/黑龙江省植物抗性研究中心,黑龙江大庆 163000)
【】检测黑龙江省稻瘟病菌()无毒基因和在不同地区、年份间流行菌株的分布情况与变异机制,了解其等位基因的致病表型,为黑龙江省抗瘟品种布局提供参考依据。利用NCBI中公布的无毒基因序列对3个无毒基因和的基因全长和编码序列(coding sequence,CDS)分别设计特异性引物,将2016年和2017年采自黑龙江省不同地区335个稻瘟病菌单孢分离菌株的DNA进行PCR扩增,通过琼脂糖凝胶电泳检测分析,并挑选不同带型和不同地区代表菌株的PCR产物进行测序。测序结果与相应无毒基因序列进行碱基与氨基酸序列的比较分析,并利用水稻抗性单基因系,对不同变异类型的稻瘟病菌菌株进行致病型测定。在PCR电泳检测中和均出现特异性条带,说明这3个无毒基因在黑龙江省均有分布,并以不同分布频率与突变类型出现。3个无毒基因平均扩增频率分别为75.52%、87.16%和85.67%。其中通过电泳检测与PCR产物测序检测出4种带型(无带、高带、中高带与低带)和5种变异类型(1-1、1-2、2、3、3-1),基因型1-1、1-2、-2和-3-1为新发现的变异类型,其中基因型1-1和1-2均为转座子Pot2的插入,但插入位点不同;基因型-2在阅读框上游存在小片段的插入;基因型-3-1碱基序列与原序列比对有4处差异,即32(C/G)35(T/A)36(T/A)38(T/A),导致氨基酸翻译提前终止。对检测到的5种等位基因型进行致病型测定,分析发现除正常基因型3外,其他变异基因型无毒功能均已丧失。而经PCR产物测序检测出7种等位基因型,(D、A、B、C、E、F、F2),碱基序列的变化均导致氨基酸错义突变,7种等位基因型均已见报道。无毒基因通过电泳检测和PCR产物测序分析,发现2种带型(高带和正常带型)与4种基因型(A、B、C、D),其中A为正常基因型;基因型B在191位处有碱基A的插入,导致氨基酸翻译提前终止;基因型为新发现的等位基因型,其特点在于与A类型存在17(T/C)和19位处碱基C的插入,发生移码突变翻译提前终止;高带型对应的无毒基因型D经测序验证为Pot3转座子的插入。4种基因型菌株分别接种水稻单基因系发现,A可以被识别表现无毒,(B、C、D)均不能被所识别而表现为有毒性。黑龙江省稻瘟病菌无毒基因和分布较为广泛,且变异类型丰富,研究结果可为选育和推广携带相应抗病基因的水稻品种提供参考。
黑龙江省;稻瘟病菌;无毒基因;;;
0 引言
【研究意义】稻瘟病菌()引起的稻瘟病是世界水稻生产中最具有毁灭性的病害之一[1-2],目前,控制该病害最为经济有效并且对环境安全的策略是种植抗病品种。但经常一个高抗水稻品种由于病原菌无毒基因的突变或者有毒群体的增长,导致抗病品种应用3—5年后便“丧失”抗性,变为高度的感病品种。因此,对水稻与稻瘟病菌的互作调控机制深入研究,了解病原物丧失无毒功能的机制并阐明无毒基因变异对水稻抗性的影响,从而进一步了解无毒基因和对应抗性基因之间的互作关系,对长久、有效地防治稻瘟病具有重要意义。【前人研究进展】稻瘟病系统是一个经典的基因对基因系统[3],其中致病菌中的无毒(AVR)基因在功能上与水稻中的特定抗病基因(R)相对应。只有在稻瘟病菌的无毒基因与寄主的抗性基因相互作用时,水稻品种才能表现出抗瘟性[4-5]。近年来在水稻抗瘟基因与稻瘟病菌无毒基因研究方面已经取得了非常重要的研究进展,据不完全统计,目前已鉴定的无毒基因有40多个[6],其中、和等被克隆[7-16]。无毒基因在病菌群体中的存在、缺失及变异会影响与寄主植物抗性基因的识别,对于病菌群体中无毒基因的研究有利于抗病品种的选育和利用。李祥晓等[17]对177个采自黑龙江省稻瘟病菌单孢菌株进行PCR扩增检测,并采用20个已知抗性基因单基因系对48个黑龙江省稻瘟病菌进行毒力分析,结果表明黑龙江省稻瘟病菌无毒基因具有明显的地理特征和品种的特异性,在不同地区应根据无毒基因组成选取有效的抗病基因用于水稻抗瘟育种;王世维等[18]选取辽宁省稻瘟病常发区的26株稻瘟病菌单孢菌株,对6个无毒基因的PCR产物进行碱基与氨基酸序列比较分析,结果表明不同地区稻瘟病菌菌株的无毒基因类型及发生频率差异较大,可为辽宁省不同稻区的品种种植提供参考依据;汪文娟等[19]采用抗稻瘟病单基因系对稻瘟病菌单孢分离菌株进行致病型测定,并根据已经克隆的且与稻瘟病菌致病性相关的8个无毒基因的功能性分子标记进行分离菌株的无毒基因型分析,结果表明在华南主栽品种广8 A杂交稻组合中分布较为广泛的无毒基因有和。【本研究切入点】深入阐明稻瘟病菌无毒基因的变异特征,需要从等位基因型与病原菌致病型两方面开展研究,据前人报道抗性基因和在黑龙江省主栽水稻品种中存在较为广泛,并且具有较高的利用价值[20-21]。为准确探明黑龙江省稻瘟病菌无毒基因、和的分布与变异机制,本研究结合无毒基因的扩增与测序结果和抗病单基因系品种致病型鉴定,对采集自黑龙江省的稻瘟病菌进行分析,研究不同菌株中无毒基因的类型及变异情况。【拟解决的关键问题】通过基因扩增,首先判断不同地区的稻瘟病菌是否携带无毒基因或其等位基因,再通过测序比对分析菌株之间无毒基因序列的变化,最后通过对不同变异类型的稻瘟病菌单孢分离菌株进行致病型测定,从而明确黑龙江省稻瘟病菌无毒基因分布情况及变异类型,以期为黑龙江省抗瘟品种的合理布局与稻瘟病的有效防控提供依据。
1 材料与方法
试验于2018年6月至2019年4月在黑龙江八一农垦大学植物抗性研究中心实验室完成。
1.1 供试菌株
2016年和2017年在黑龙江省10个市20个县水稻种植区内采集稻瘟病穗颈瘟标样,经单孢分离获得单孢菌株335个(2016年菌株127个,2017年菌株208个),采用滤纸片保存法[22]保存备用。
1.2 稻瘟病菌基因组DNA提取
将分离纯化的稻瘟病菌单孢菌株在PDA固体培养基上活化培养,挑取适量菌丝块接到酵母液体培养基中,于28℃摇床,120 r/min振荡培养3—5 d,收集菌丝体,分装于1.5 mL离心管中,置于-20℃冰箱中冷冻保存。使用真菌DNA提取试剂盒(D3390-01 OmegaBio-Tek公司)提取稻瘟病菌基因组DNA,DNA提取之后使用微量分光光度计测定DNA浓度,并将DNA原液稀释成60 ng·µL-1的工作液备用。原液-20℃保存。
1.3 引物设计
根据文献中已克隆的无毒基因于NCBI上查找基因序列,利用Primer premier 5.0在基因全长和CDS区设计6对特异性引物,所有引物均委托上海生工生物工程技术有限公司合成,引物序列见表1。

表1 用于扩增稻瘟病菌无毒基因的引物
F:正向引物 Forward primer;R:反向引物Reverse primer
1.4 PCR扩增及电泳检测
PCR反应体系(20 µL):rTaq酶(5 U·μL-1)0.1 µL,10×Buffer(Mg+)2.0 µL,dNTP Mixture 1.6 µL(2.5 m Meach),正反向引物(10 μmol·L-1)各0.3 µL,DNA模板(20—30 ng·μL-1)1 µL,加ddH2O补足20 µL。扩增程序:94℃预变性4 min,94℃变性45 s,55—60℃退火45 s(具体温度根据引物的GC含量设定),72℃延伸30—90 s(具体时间根据扩增片段大小确定),30个循环,72℃延伸10 min,4℃保存待检测。扩增产物在1%的琼脂糖凝胶中电泳,150 V,20 min,电泳液为0.5×TBE,在凝胶电泳成像系统下观察并拍照,统计无毒基因扩增频率。
1.5 部分无毒基因序列测序分析
在黑龙江省2016年和2017年的稻瘟病菌菌株中挑选不同带型与不同地区的部分菌株送去上海生工生物工程技术有限公司测序,测序结果采用Lasergene 7.0的SeqMan软件进行比对与拼接;并采用DNAMAN软件对有差异的核苷酸序列进行比较分析。
1.6 致病型测定
植物材料:2个抗稻瘟病单基因系分别为BL1()和IRBLzt-T(),均为国际水稻研究所选育,感病对照:丽江新团黑谷(LTH)。
菌株活化及产孢培养:在无菌条件下,将之前保存的滤纸片置入PDA培养基内培养,5 d后转置米糠培养基内进行产孢培养。当培养皿内长满菌丝后,将气生菌丝用涂布棒刮掉,将其放置在恒温恒湿环境中,用蓝紫光灯照射3 d,进行产孢培养。
植物材料种植:水稻品种采用室内水培方式进行培养。挑选饱满的种子用1.25‰的多菌灵溶液室温(20℃)浸泡24 h后,用清水冲洗干净,放入培养皿内并用滤纸保湿。于30℃的恒温培养箱内催芽,播种于固定在塑料水培盒(尺寸:225 mm×155 mm×55 mm)内的定植篮(上端内径34 mm,下端内径28 mm,高45 mm)中,每个定植篮内播种10粒,自来水培养3 d以后,换营养液培养,营养液采用经典的霍格兰(Hoagland)配方[23],每4 d换一次营养液,待苗长至3叶1心期进行人工喷雾接种。
接种及调查:每个培养皿用5 mL无菌水洗下孢子,用双层纱布过滤,经显微镜检测当孢子量为1×105个/mL时,将菌悬液装入喷壶中,并加入5 mL明胶溶液摇匀后均匀地喷洒在秧苗表面,接种后将其移入25℃的保湿棚内进行遮光保湿培养24 h,然后返回室温自然正常培养,接种后7 d调查,调查标准参考靳学慧等[24]。
2 结果
2.1 稻瘟病菌无毒基因AVR-Pib的基因型与致病表型分析
以稻瘟病菌335个菌株的DNA为模板,根据无毒基因外显子区域与全序列设计的引物(图1)进行PCR扩增,电泳结果显示有4种不同类型的条带(图2),分别为无带、高带、中高带与低带,用0、1、2、3表示,带型0含有82个菌株,带型1含有8个菌株,带型2为2个,带型3的菌株有243个。将包含带型1、2、3的稻瘟病菌菌株共40株的PCR产物进行测序(其中带型1的菌株4个;带型2的菌株2个;带型3的菌株34个)。测序结果发现4个新的基因型,即带型1的菌株一个基因型是在基因阅读框上游-37 bp处有转座子Pot2的插入,另一个基因型是阅读框的5′端26 bp处有转座子Pot2的插入,将其分别命名为1-1和1-2。带型2的菌株碱基序列在基因上游-129 bp处有500 bp的碱基序列插入,将其命名为2;带型3的菌株YL17077序列比对有4处差异,即32(C/G)35(T/A)36(T/A)38(T/A)(图3),导致翻译提前终止,将其命名为3-1,其他菌株的序列均与序列完全一致,且出现频率最高,命名为-3。

图1 无毒基因AVR-Pib两个引物AVR-Pib F1/R1和AVR-Pib F2/R2的位置
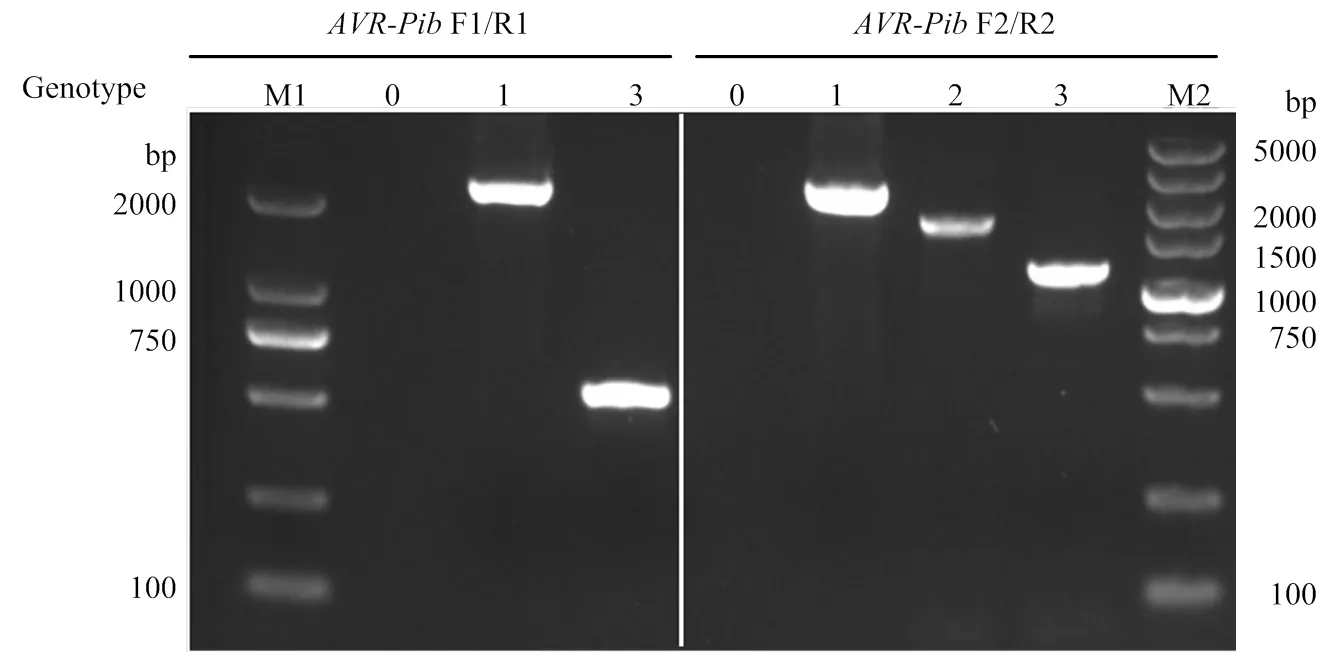
M1:Marker,DL2000;M2:Marker,DL5000;0:未扩增出条带no band;1:高带型基因high band alleles;2:中高带型基因 Mid to high band alleles;3:低带型基因(正常基因)lower band alleles (general gene)
采用--3及4种变异类型--1-1、--1-2、--2和--3-1的菌株接种水稻单基因系BL1(),以丽江新团黑谷(LTH)为对照。结果显示正常基因型菌株能够诱导产生无毒性(A),而4种新变异类型均不能被识别而表现为有毒性(V),表明此变异类型造成无毒功能丧失(图4)。
2.2 稻瘟病菌无毒基因AVR-Pik的扩增结果与序列分析
以黑龙江省稻瘟病菌335个菌株的菌丝DNA为模板,根据无毒基因-D序列设计引物扩增,电泳结果显示有292株可扩增出特异性片段。从292株菌株中挑选不同地区与不同年份的菌株40株进行测序,与NCBI中-D的CDS序列比对分析,结果发现7种等位基因型,且均有文献报道[13,18,25]。其中有6个菌株序列与报道的-D序列完全一致;其余菌株的PCR产物序列与已经克隆的-D序列有着不同程度的差异(图5)。根据差异可分为6种类型:第1类,与Yoshida等[13]文中报道的-A类型序列一致,与-D相比有3处差异,即136(C/A)、139(C/G)和143(G/A);第2类,与Yoshida等[13]文中报道的-B类型序列一致,与-D相比有4处差异,即136(C/A)、139(C/G)、143(G/A)和234(G/A);第3类,与Yoshida等[13]文中报道的-C类型序列一致,与-D相比有2处差异,即136(C/A)和200(C/A);第4类,与Yoshida等[13]文中报道的-E类型序列一致,与-D相比有1处差异,即136(C/A);第5类,与王世维等[18]文中新增的-F类型序列完全一致,与-D相比有1处差异,即143(G/A);第6类,与Longya等[25]文中报道的新基因型-F序列一致,与-D相比有4处差异136(C/A)、139(C/G)、143(G/A)和234(T/A),本试验将其命名为-F2。
6类基因型碱基序列翻译的氨基酸序列与基因组(D型)的氨基酸序列比对结果如图6。-A的氨基酸序列存在3处错义突变,分别为46(H/N)、47(P/A)和48(G/D);-B的氨基酸序列存在4处错义突变,分别为46(H/N)、47(P/A)、48(G/D)和78(M/I);-C的氨基酸序列存在2处错义突变,分别为46(H/N)和67(A/D);-E的136(C/A)SNP造成了氨基酸突变46(H/N);-F的143(G/A)SNP造成了氨基酸突变48(G/D)。-F2的氨基酸序列存在3处错义突变46(H/N)、47(P/A)、48(G/D)和78(M/K)。
2.3 稻瘟病菌无毒基因AvrPiz-t的基因型与致病表型分析
以黑龙江省稻瘟病菌335个菌株的菌丝DNA为模板,根据无毒基因的外显子区域与基因全长设计引物扩增,电泳结果显示有287株可扩增出特异性片段(图7),有4个菌株扩增出高带型。从中挑选40个不同带型、不同地区有特异性扩增片段的稻瘟病菌菌株PCR产物进行测序,结果发现4种基因型:第1类,菌株序列与基因原序列相同将其命名为A;第2类,在CDS的191 bp处有碱基A的插入,本试验将此变异类型命名为B;第3类,与A存在2处差异,即CDS的17 bp处碱基C的替换和19 bp处碱基C的插入,此变异类型命名为C(图8);第4类,在启动子区域-462 bp处有转座子Pot3插入,此变异类型已有报道且无毒功能已丧失[12],本试验将其命名为D(图9)。对推测的氨基酸序列进行比较分析发现,B在64位发生移码突变,导致氨基酸序列至89位时提前终止;C在第6位发生移码突变,并在46位处提前终止(图10)。

图3 AVR-Pib等位基因变异的特征

A:无毒性avirulent;V:有毒性virulent

图5 供试菌株的AVR-Pik 碱基序列比对

图6 AVR-Pik氨基酸序列对比分析
对无毒基因型突变类型的致病型进行了分析,如图11所示,以丽江新团黑谷(LTH)为对照,结果显示,A可以被识别表现无毒性,B、C和D均不能被识别而表现为有毒性。

a:AvrPiz-t F1/R1的扩增结果amplification result of AvrPiz-t F1/R1;b:AvrPiz-t F2/R2的扩增结果amplification result of AvrPiz-t F2/R2。M1:Marker,DL2000;M2:Marker,DL5000;“+”:正常基因型Normal genotype;“++”:高带型High band type;“-”:未扩增出条带no amplification band;“guy11”:标准菌株Standard strain

图8 供试菌株AvrPiz-t碱基序列比对

图9 AvrPiz-t特点及引物设计

图10 AvrPiz-t氨基酸序列对比分析
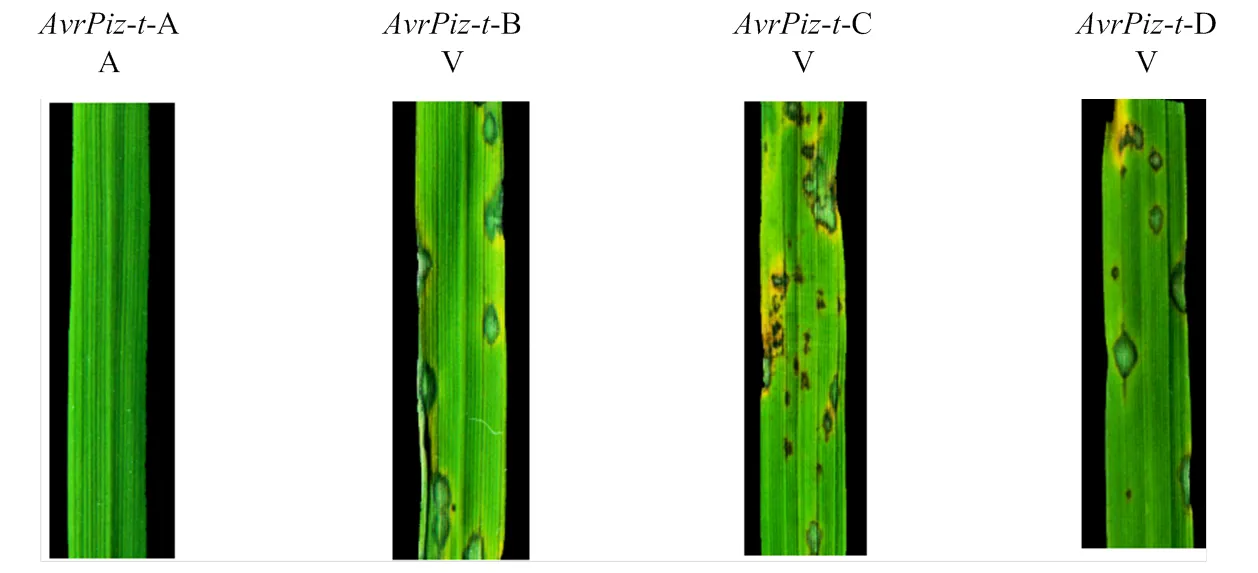
A:无毒性avirulent;V:有毒性virulent
2.4 3种无毒基因在黑龙江省的分布情况
对2016年和2017年黑龙江省335个稻瘟病菌单孢菌株3个无毒基因、和的检测结果显示,这3个无毒基因在黑龙江省各稻区以不同分布频率与不同等位基因类型出现,连续两年出现频率较高且稳定遗传,平均出现频率均超过70%,分别为75.52%、87.16%和85.67%(表2)。表明这3个无毒基因为黑龙江省稻瘟病菌携带的重要无毒基因。

表2 2016年和2017年黑龙江省稻瘟病菌无毒基因的扩增频率
a:2016年参试菌株数 Number of test strains in 2016;b:2017年参试菌株数Number of test strains in 2017;c:总参试菌株数 Total number of test strains
3 讨论
3.1 黑龙江省稻瘟病菌的无毒基因型与品种的合理布局
黑龙江省是中国北方粳稻主产区,种植面积占东北地区总面积的58.1%[26],由于抗性品种抗性“丧失”使稻瘟病危害日益加重而造成粮食的大面积减产甚至绝收。稻瘟病菌的无毒基因可与相应的抗性基因互作,导致水稻植株发生非亲和反应而产生抗性,因此,监测黑龙江省主要水稻种植区稻瘟病菌的无毒基因型,合理利用抗病品种可以很大程度上降低稻瘟病发生。本试验检测到、和这3种无毒基因在黑龙江省稳定存在,无毒基因型-3和-A出现频率最高且在黑龙江省各稻区均有分布,根据本试验的致病型测定来看,-3对抗性品种表现为无毒性。据时克等[27]报道在中国水稻品种中锦辉501等9个籼稻品种和粳稻品种辽粳454、武育粳7号携带抗性基因,刘华招等[20]在黑龙江省种植品种中检测到垦稻13、垦稻14、松粳9、龙粳2这4份材料含有。所以在品种选育时可以种植以上携带的水稻品种。-A对表现出无毒性,据于连鹏[21]报道黑龙江省48个主栽水稻品种中含有的占77.08%且分布较广,因此携带抗性基因的品种在黑龙江省抗病育种和品种合理布局时可以选用。在的等位基因中,F2与C出现频率最高,据文献报道F2和C均不能被抗性基因、和所识别[25,28],因此在生产上要避免利用携带、或的品种。
3.2 无毒基因的变异机制与功能验证
病原菌无毒基因不稳定,常常引发变异,无毒基因的变异导致其逃脱寄主抗病基因的识别使抗病品种丧失抗病性。因此,了解无毒基因变异机制对于培育抗病品种是至关重要的。无毒基因的变异机制主要包括点突变、缺失、插入、复制、移码突变等[29]。
变异能力较强,据Zhang等[30]报道检测出5种带型(无带、高带、中高带、低带与双带)和11种变异类型,且13位处的异亮氨酸为热点可变位置。在本研究中,黑龙江省稻瘟病菌检测出4种带型(无带、高带、中高带与低带)和5种基因型,未检测到双带型,变异类型-3-1在热点可变位置翻译提前终止,由于此变异类型会导致抗性基因的功能丧失,所以应引起研究者的关注。
与等位基因间呈现出了竞争性阶梯型共进化模式[31],据Kanzaki等[28]研究报道,与具有高度变异性,主要突变类型是编码区单碱基的突变从而引起氨基酸的变异,5个氨基酸突变活性位点为46、47、48、67和78[13]。本试验检测出7种等位基因型,且无毒基因序列变异会导致其毒性发生变化。因此如何使用抗谱更广的抗性基因或采用多基因联合手段克服的变异仍需进一步的研究。
是一个片段长度为327 bp,无内含子的基因,编码一个含有108个氨基酸的小分泌蛋白。它存在于有毒菌株guy11中,但在其启动子区域-462 bp处有转座子pot3的插入,使基因表达不稳定无毒功能丧失[11]。的变异类型主要有:一是插入突变,如在启动子区域和ORF区域插入片段大小不等的重复序列,如转座子Pot2、Pot3,逆转录转座子Inago2、MGR583等。二是点突变,如编码区域第41位氨基酸的改变(A41V)[32]。本试验中插入突变和点突变均有发现。其中B曾在2014和2015年黑龙江省稻区内被发现[33],但当时是否丧失无毒功能未被验证。C的变异类型为点突变和单个碱基的插入,导致翻译提前终止,D为-462 bp处插入pot3转座子,通过基因功能验证得知(B、C、D)已丧失无毒功能。
4 结论
无毒基因和在黑龙江省均有分布,并以不同分布频率与不同变异类型出现,且3种无毒基因型在黑龙江省稻瘟病菌中稳定遗传。无毒基因在黑龙江省检测出4种带型(无带、高带、中高带与低带),同时检测出5种基因型(1-1、1-2、2、3、3-1),其中(1-1、1-2、2、3-1)无毒功能均已丧失;检测出7种等位基因型,(D、A、B、C、E、F、F2),均有文献报道;本试验检测出4种基因型(A、B、C、D),C为新增基因型,(B、C、D)失去无毒功能。黑龙江省各稻区稻瘟病菌复杂多样,但能够通过及时地监测无毒基因在稻瘟病病原菌群体中的组成与变化,来指导田间抗性基因的合理布局和抗病品种的定期轮换,以有效地控制病害发生,并为抗病种质资源筛选提供参考依据。
[1] Couch B C, Kohn L M. A multilocus gene genealogy concordant with host preference indicates segregation of a new species,, from., 2002, 94(4): 683-693.
[2] Ou S H.. Kew, UK: Commonwealth Mycological Institute, 1985.
[3] Flor H H. Current status of the gene-for-gene concept., 1971, 9: 275-296.
[4] Marcel S, Sawers R, Oakeley E, Angliker H,Paszkowski U. Tissue-adapted invasion strategies of the rice blast fungus., 2010, 22(9):3177-3187.
[5] Liu W, Zhou X, Li G, Li L, Kong L, Wang C, Zhang H, Xu J R. Multiple plant surface signals are sensed by different mechanisms in the rice blast fungus for appressorium formation., 2011, 7(1): e1001261.
[6] Ma J H, Wang L, Feng S J, Lin F, Xiao Y, Pan Q H. Identification and fine mapping of, a novel avirulence gene of., 2006, 113(5): 875-883.
[7] Sweigard J A, Carroll A M, Kang S, Farrall L, Chumley F G, Valent B. Identification, cloning, and characterization of, a gene for host species specificity in the rice blast fungus., 1995, 7(8):1221-1233.
[8] Kang S, Sweigard J A, Valent B. Thehost specificity gene family in the blast fungus., 1995, 8(6):939-948.
[9] Orbach M J, Farrall L, Sweigard J A,Chumley F G, Valent B. A telomeric avirulence gene determines efficacy for the rice blast resistance gene., 2000, 12(11):2019-2032.
[10] Collemare J, Pianfetti M, Houlle A E,Morin D, Camborde L, Gagey M J, Barbisan C, Fudal I, Lebrun M H, BőHnert H U.avirulence genebelongs to an infection-specific gene cluster involved in secondary metabolism., 2008, 179(1):196-208.
[11] Farman M L, Leong S A. Chromosome walking to theavirulence gene of: discrepancy between the physical and genetic maps., 1998, 150(3):1049-1058.
[12] Li W, Wang B, Wu J, Lu G,Hu Y, Zhang X, Zhang Z G, Zhao Q, Zhang H Y, Wang Z Y, Wang G L, Han B, Wang Z H, Zhou B. Theavirulence geneencodes a predicted secreted protein that triggers the immunity in rice mediated by the blast resistance gene., 2009, 22(4):411-420.
[13] Yoshida K, Saitoh H, Fujisawa S, Kanzaki H, Matsumura H, Yoshida K, Tosa Y, Chuma I, Takano Y, Win J, Kamoun S, Terauchi R. Association genetics reveals three novel avirulence genes from the rice blast fungal pathogen., 2009, 21(5):1573-1591.
[14] Wu J, Kou Y, Bao J,Li Y, Tang M, Zhu X, Ponaya A, Xiao G, Li J, Li C, Song M Y, Cumagun C J, Deng Q, Lu G, Jeon J S, Naqvi N I, Zhou B. Comparative genomics identifies theavirulence effectorthat triggers-mediated blast resistance in rice., 2015, 206(4):1463-1475.
[15] Schneider D R, Saraiva A M, Azzoni A R, Miranda H R, de Toledo M A, Pelloso A C, Souza A P. Overexpression and purification of, a mutant of the effector protein PWL2 from., 2010, 74(1):24-31.
[16] Zhang S, Wang L, Wu W, He L, Yang X, Pan Q. Function and evolution ofavirulence generesponding to the rice blast resistance gene., 2015, 5:11642.
[17] 李祥晓,王倩,罗生香,何云霞,朱苓华,周永力,黎志康. 黑龙江省稻瘟病菌无毒基因分析及抗病种质资源筛选. 作物学报, 2012, 38(12): 2192-2197.
Li X X, Wang Q, Luo S X, He Y X, Zhu L H, Zhou Y L, Li Z K. Analyzing avirulence genes offrom Heilongjiang province and screening rice germplasm with resistance to blast fungus., 2012, 38(12): 2192-2197. (in Chinese)
[18] 王世维, 郑文静, 赵家铭, 魏松红, 王妍, 赵宝海, 刘志恒. 辽宁省稻瘟病菌无毒基因型鉴定及分析.中国农业科学, 2014, 47(3): 462-472.
Wang S W, Zheng W J, Zhao J M, Wei S H, Wang Y, Zhao B H, Liu Z H. Identification and analysis ofavirulence genes in Liaoning province., 2014, 47(3): 462-472. (in Chinese)
[19] 汪文娟, 苏菁, 杨健源, 韦小燕, 陈凯玲, 陈珍, 陈深, 朱小源. 源于广8 A杂交稻组合的稻瘟病菌无毒基因型分析. 中国农业科学, 2018, 51(24): 4633-4646.
Wang W J, Su J, Yang J Y, Wei X Y, Chen K L, Chen Z, Chen S, Zhu X Y. Analysis ofavirulent genes in the infected hybrid rice combinations derived from a sterile line of guang 8 A., 2018, 51(24): 4633-4646. (in Chinese)
[20] 刘华招, 刘延, 刘化龙, 徐正进, 陈温福. 黑龙江省种植品种中稻瘟病抗性基因和的分布. 东北农业大学学报, 2011, 42(4): 27-31.
Liu H Z, Liu Y, Liu H L, Xu Z J, Chen W F. Distribution of two rice blast resistance genesandin major rice cultivars in Heilongjiang province in China., 2011, 42(4): 27-31. (in Chinese)
[21] 于连鹏. 黑龙江省主栽水稻品种、和抗瘟基因检测和抗性评价[D]. 大庆: 黑龙江八一农垦大学, 2017.
Yu L P.,andgenes detection and blast resistance evaluation of main rice varieties in Heilongjiang province[D]. Daqing: Heilongjiang Bayi Agricultural University, 2017. (in Chinese)
[22] 蒋金芬, 韩红萍, 梁友方. 滤纸片法低温冷冻保存菌种的实验室应用. 中国公共卫生, 2006, 22(3): 310.
Jiang J F, Han H P, Liang Y F. Laboratory application of filter paper method for cryopreservation., 2006, 22(3): 310. (in Chinese)
[23] 连兆煌. 无土栽培原理与技术. 北京: 农业出版社, 1994.
Lian Z H.. Beijing: Agriculture Press, 1994. (in Chinese)
[24] 靳学慧, 马汇泉. 农业植物病理学. 赤峰: 内蒙古科学技术出版社, 1999.
Jin X H, MA H Q..Chifeng: Inner Mongolia Science and Technology Press, 1999. (in Chinese)
[25] Longya A, Chaipanya C, Franceschetti M, Maidment J H.R, Banfield M J, Jantasuriyarat C. Gene duplication and mutation in the emergence of a novel aggressive allele of theeffector in the rice blast fungus., 2019, 32(6): 740-749.
[26] 孙强, 张三元, 张俊国, 杨春刚. 东北水稻生产现状及对策. 北方水稻, 2010, 40(2): 72-74.
Sun Q, Zhang S Y, Zhang J G, Yang C G. Current situation of rice production in northeast of china and countermeasures., 2010, 40(2): 72-74. (in Chinese)
[27] 时克, 雷财林, 程治军, 许兴涛, 王久林, 万建民. 稻瘟病抗性基因和在我国水稻主栽品种中的分布.植物遗传资源学报, 2009, 10(1): 21-26.
Shi K, Lei C L, Cheng Z J, Xu X T, Wang J L, Wan J M. Distribution of two blast resistance genesandin major rice cultivars in China., 2009,10(1):21-26.(in Chinese)
[28] Kanzaki H, Yoshida K, Saitoh H, Fujisaki H, Hirabuchi A, Alaux L, Fournier E, Tharreau D, Terauchi R. Arms race co-evolution ofand ricegenes driven by their physical interactions., 2012, 72(6): 894-907.
[29] 姜华, 余欢, 王艳丽, 孙国昌. 稻瘟病菌无毒基因序列变异研究进展.浙江农业学报, 2015, 27(3): 512-520.
Jiang H, Yu H, Wang Y L, Sun G C. Progress on sequence variation of avirulence gene in the rice blast fungus., 2015, 27(3): 512-520. (in Chinese)
[30] Zhang S, Wang L, Wu W, He L, Yang X, Pan Q. Function and evolution ofavirulence generesponding to the rice blast resistance gene., 2015, 5: 11642.
[31] Wu W H, Wang L, Zhang S, Liang Y Q, Zheng X L, Yi K X, He C P. Assessment of sensitivity and virulence fitness costs of thealleles fromto isoprothiolane., 2014, 13(4): 9701-9709.
[32] 陈美莲. 稻瘟病菌无毒基因的遗传变异分析[D]. 福州: 福建农林大学, 2014.
Chen M L. Genetic variation of avirulence genein[D]. Fuzhou: Fujian Agriculture and Forestry University, 2014. (in Chinese)
[33] 刘殿宇. 黑龙江省稻瘟病菌致病性与无毒基因检测及遗传多样性分析[D]. 大庆: 黑龙江八一农垦大学, 2017.
Liu D Y. Pathogenicity avirulent gene detection and genetic diversity analysis ofin Heilongjiang province[D]. Daqing: Heilongjiang Bayi Agricultural University, 2017. (in Chinese)
Detection and analysis ofAvirulence Genesandin Heilongjiang province
MENG Feng, ZHANG YaLing, JIN XueHui, ZHANG XiaoYu, JIANG Jun
(College of Agronomy, Heilongjiang Bayi Agricultural University/Heilongjiang Plant Resistance Research Center, Daqing 163000, Heilongjiang)
【】The objective of this study is to investigate the distribution and variation mechanism of avirulence genes,andinstrains from different regions and years in Heilongjiang Province, to understand the pathogenic phenotypes of different avirulence gene alleles, and to provide a reference for utilization and distribution of resistance cultivars in Heilongjiang Province.【】Based on the avirulence gene sequences published in NCBI, specific primers were designed to amplify full length and the coding sequence (CDS) regions of three genes, respectively. From 2016 to 2017, 335strains in different regions of Heilongjiang Province were collected and isolated, and their DNA was PCR-amplified using avirulence genes primers and analyzed by agarose gel electrophoresis. the PCR products with different band patterns and from representative strains of different regions were selected for sequencing. The sequencing results were compared with the corresponding avirulence gene sequences for base and amino acid. The pathogenic phenotype ofstrains with different variant types was determined based on the rice resistance to single-gene lines.【】The specific bandsof,andwere detected in PCR detection and appeared in different distribution frequencies and mutation types, indicating that these 3 avirulence genes were all distributed in Heilongjiang Province. The average amplification frequency of the 3 avirulence genes was 75.52%, 87.16% and 85.67%, respectively. Among them, 4 types of band (bandless, high band, mid to high band and low band) ofwere detected by electrophoresis analysis and 5 variant types(1-1, 1-2, 2, 3, 3-1) were detected by PCR product sequencing. The genotypes1-1,1-2,-2 and-3-1 are newly discovered variant types, of which genotypes-1-1 and1-2 are insertions of transposon Pot2 but with different insertion sites. The genotype2 has a small fragment insertion in the upstream of CDS region. The genotype3-1 base sequence has 4 differences from the original sequence, namely 32 (C/G) 35 (T/A) 36 (T/A) 38 (T/A), and the amino acid translation was terminated prematurely. Pathogenic analysis showed that except for the normal genotype3, the other alleles lost their avirulence functions. Sevenalleles (D, A, B, C, E, F, F2) were detected after PCR product sequencing, and the alterations in the nucleotide sequences of these alleles all resulted in amino acid missense mutations. The 7alleles have been reported previously. The avirulence genewas analyzed by electrophoresis and sequencing of PCR products, and 2 types of band (high band and normal band type) and 4 genotypes of(A, B, C, D) were revealed.Among them,-A is the original genotype, while-B has a base A insertion at position 191, causing premature termination of amino acid translation. Genotype-C is a newly discovered allelic type, characterized by the presence of a nucleotide variation at position 17 (T/C) and the insertion of base C at position 19 compared with type A, leading to the frameshift mutation and premature translation termination. The high band type avirulent genotype-D was sequenced and verified as having an insertion of the pot3 transposon. Rice single-gene lines infection showed that the strains with-A were avirulent toline due torecognition, whereas the strains with(B, C, D) were virulent toline due to lost the ability recognized by.【】The avirulence genes,andofin Heilongjiang province are widely distributed and the types of variation are abundant. The results of this study can provide a reference for breeding and popularizing rice cultivars with corresponding disease-resistance genes.
Heilongjiang Province;; avirulence gene;;;

10.3864/j.issn.0578-1752.2019.23.007
2019-05-30;
2019-08-01
黑龙江省自然科学基金(QC2011C046)、黑龙江八一农垦大学学成、引进人才科研启动计划(XDB-2016-05)、黑龙江省农垦总局科技攻关项目(HNK125A-08-06,HNK135-02-02)、黑龙江省教育厅项目(12521376)
孟峰,E-mail:xmfengf@126.com。
张亚玲,E-mail:byndzyl@163.com。通信作者靳学慧,E-mail:Jxh2686@163.com
(责任编辑 岳梅)
