RP-HPLC法测定植物油中苯并(a)芘残留量
2020-01-13邹沫君
邹沫君
乐山市食品药品检验检测中心(乐山 614000)
苯并(a)芘是一种常见的高活性间接致癌物和突变原,必须经细胞微粒体中的混合功能氧化酶激活才具有致癌性[1]。食品中苯并芘化合物主要来源于:熏烤或高温烹调时使食品污染苯并芘;食品加工过程中被污染,如沥青污染、包转材料污染、环境污染等[2]。可通过改进食品加工方法,如熏制和烘烤食品时,改进燃烧过程,避免食品直接接触炭火,改进熏烟工艺等措施降低污染[3]。试验采用简单二元流动相,以等度洗脱提高分离效能,试样经正己烷提取,Cleanert BAP净化,浓缩至干后经乙腈复溶,反相液相色谱分离,荧光检测器检测,建立了测定植物油中苯并芘的品质控制分析方法,并对市面上20个不同厂家植物油中苯并芘残留量进行比较,旨在更好地控制植物油的内在品质,保证食用植物油的安全、优质。
1 仪器与材料
1.1 仪器与设备
DGU-20A 3R高效液相色谱仪(带RF-20A荧光检测器,日本岛津公司);FA124电子天平(上海舜宇恒平科学仪器有限公司);TG-16医用离心机(四川蜀科仪器有限公司);XH-D旋涡混合器(上海皓庄仪器有限公司);JHD-002干式氮吹仪(上海极恒实业有限公司);KQ-700型超声波清洗器(昆山市超声仪器有限公司);Cleanert BAP(500 mg/6 mL 30/pkg,Agela Technologies)。
1.2 材料与试剂
20批试样(市售,包括10批次压榨工艺植物油、10批次浸出工艺植物油,每批试样均来自不同的生产厂家);苯并(a)芘标准品(100 μ g/mL in Acetonitrile,98.8%,1 mL,Lot. 170317,0~8 ℃ and dark,BePure);乙腈、二氯甲烷、正己烷、甲醇、无水乙醇均为色谱纯(北京百灵威科技有限公司);超纯水(电阻率=18.2 MΩ·cm)。
2 方法与结果
2.1 色谱分离系统的确定
2.1.1 对照品储备液的制备
精密吸取0.05 mL苯并(a)芘标准品,用乙腈定容到250 mL,避光保存在0~5 ℃的冰箱中,保存期1个月。
2.1.2 供试品溶液的制备
称取0.4 g(精确到0.001 g)各厂家试样,加入5 mL正己烷,漩涡混合0.5 min,在40 ℃下超声提取10 min,以4 000 r/min离心5 min,转移出上清液。加入5 mL正己烷重复提取1次。合并上清液,待净化。将待净化液转移进Cleanert BAP(依次用5 mL二氯甲烷及5 mL正己烷活化柱子),待液面降至柱床时,用6 mL正己烷淋洗柱子,弃去流出液。用6 mL二氯甲烷洗脱并收集净化液到试管中。将净化液在40 ℃下氮气吹干,准确吸取2 mL乙腈,涡旋复溶0.5 min,过微孔滤膜后供液相色谱测定。同时做试样空白试验。
2.1.3 色谱条件和系统适应性试验
采用Thermo AcclaimTM120 C18色谱柱(250 mm×4.6 mm,5.0 μm);流动相,乙腈-水(90︰10,V/V);荧光检测器:激发波长384 nm,发射波长406 nm;流速1.0 mL/min;柱温35 ℃;进样量20 μ L。
取2.1.1的对照品溶液,在2.1.3的色谱条件下进行分析。苯并(a)芘拖尾因子为0.96,理论塔板数(USP)为19 692。
2.1.4 方法专属性试验
苯并(a)芘标准溶液、阴性试样溶液及典型阳性试样溶液色谱图见图1~图3。结果表明,方法专属性好,选择性高。
2.2 分析方法验证
2.2.1 线性关系、检出限和定量限考察
分别精密吸取0,0.10,0.50,1.00,2.00,5.00,10.00和20.00 μL 2.1.1的对照品溶液,注入液相色谱仪,在2.1.3的条件下测定,得到峰面积与质量浓度的线性关系,拟合回归方程式,确定相关系数、最低检出限(S/N=3)和定量限(S/N=10),详见表1。结果表明,苯并(a)芘在0~19.76 ng/mL范围内线性关系良好,完全能满足植物油中其残留量的测定。
2.2.2 精密度考察
取2.2.1的苯并(a)芘线性第4点对照品溶液,按2.1.3的色谱条件重复进样6次,记录色谱图。以峰面积计算RSD(n=6),为0.22%,表明仪器精密度良好。
2.2.3 方法重复性考察
取编号为1-1植物油试样,按2.1.2的方法制备6份供试品溶液进行色谱分析,计算残留量。苯并(a)芘残留量RSD值为0.37%,表明该方法重复性良好。
2.2.4 溶液稳定性考察
取制备好的编号为1-1植物油供试品溶液,按2.1.3的仪器条件分别于0,4,8,12,16和24 h进样测定,记录色谱峰面积,计算苯并(a)芘RSD(n=6),为0.41%,表明供试品溶液在24 h内稳定。
2.2.5 耐用性考察
取2.2.1的线性第4点对照品溶液,分别考察采用不同柱温(34,35和36 ℃),以及不同流速(0.9,1.0和1.1 mL/min)进行测定时仪器色谱行为的变化。以峰面积计,不同柱温条件下苯并(a)芘RSD(n=3)为0.58%;不同流速条件下苯并(a)芘RSD(n=3)为0.71%。表明在实际应用过程中色谱条件有微小变化时,植物油中苯并(a)芘残留量测定条件较宽,测定结果不受影响,系统具有较好的耐用性。
2.2.6 回收率考察
精密称取已测定含量的试样(编号1-1),平行9份,置于50 mL离心管中,加入适量苯并(a)芘对照品储备液,后按2.1.2和2.1.3的条件操作,结果见表2。苯并(a)芘的平均回收率为100.25%,RSD为0.62%,表明该方法具有良好回收率,准确度符合要求。
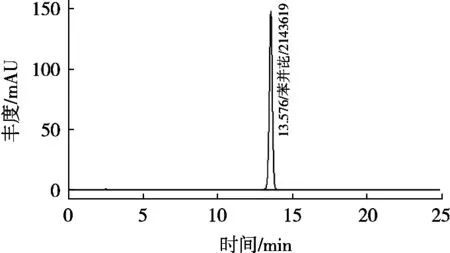
图1 苯并(a)芘标准色谱图
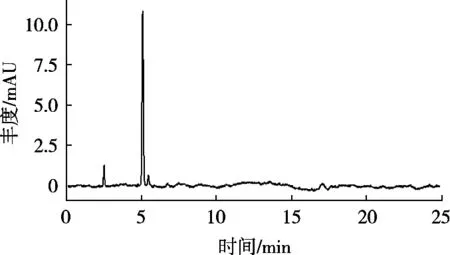
图2 阴性试样色谱图
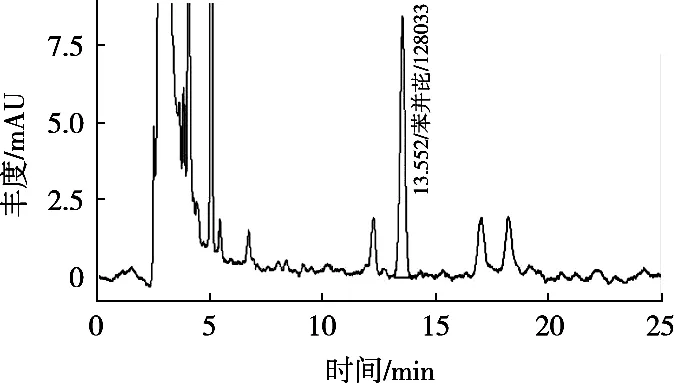
图3 典型阳性试样色谱图

表1 回归方程、相关系数、最低检出限、定量限
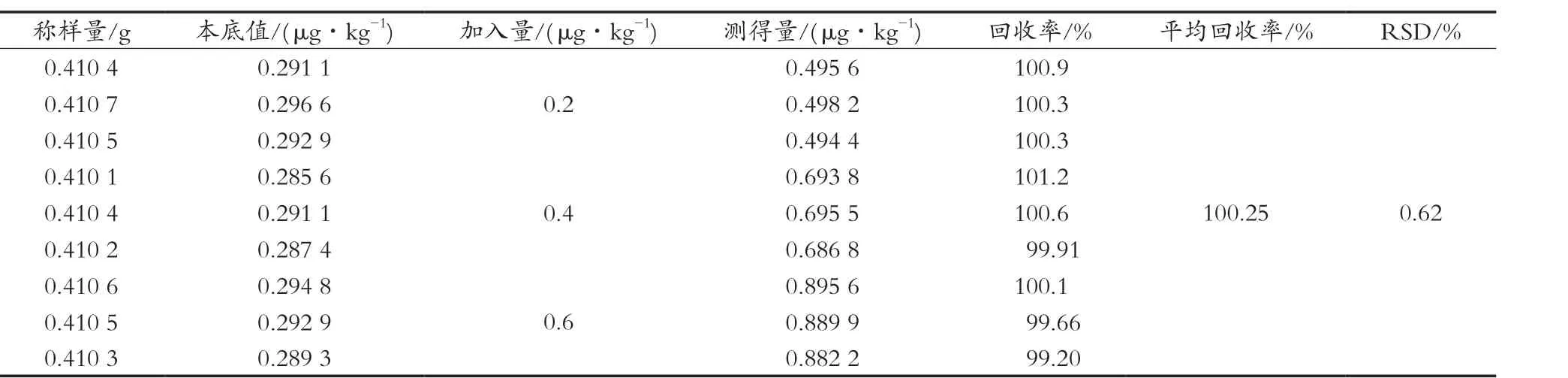
表2 回收率试验结果
2.3 样品含量测定
取20批不同厂家的植物油试样,各3份,按2.1.2的方法制备供试品溶液,在2.1.3的色谱条件下测定,采用外标法以峰面积计算含量,结果见表3。

表3 试样含量测定结果(n=3)
3 讨论
3.1 提取溶剂的选择
参考文献[4-6]苯并(a)芘残留量测定方法中供试品溶液的制备方法,考察正己烷、乙腈、甲醇、无水乙醇对苯并(a)芘提取能力的影响,发现分2次用正己烷超声处理10 min,可将样品提取完全。
3.2 流动相的选择
经查阅参考文献[7-9],最终选择乙腈-水溶液(90+10)等度洗脱,可有效缩短分析时间,保证峰形良好,适用于苯并(a)芘的大批量检测和应急检测。
3.3 含量分析
测定了20个不同生产厂家的试样,苯并(a)芘含量为0.214 7~2.883 6 μg/kg,测定结果均符合GB 2762—2017《食品安全国家标准食品中污染物限量》[10]。通过比较各组分含量测定结果,发现不同厂家间存在明显差异,其原因可能与原辅料的选取和生产工艺的控制有关。因此,食品生产者应严把原辅料进厂关,全面规范并提高生产工艺关键控制点及品质标准,杜绝勾兑现象,确保食品的安全、健康,从而更好地维护消费者舌尖上的安全。
4 结论
试验建立了植物油中苯并(a)芘残留量的RP-HPLC检测方法。该方法分离提取效果好,灵敏度、准确性高,重复性佳。可有效满足检验检测机构对植物油中苯并(a)芘残留量的测定。
