超高效液相色谱-串联质谱法定性定量检测中成药及保健食品中非法添加的17种化学药物
2020-01-10刘云杨玉忠
刘云,杨玉忠
作者单位:新乡市食品药品检验所,河南 新乡453000
消化性溃疡是临床上常见的病症,它的发作常伴有诸多的不适症状如嗳气、恶心、胃灼热等,严重影响患者的生活[1]。幽门螺杆菌感染常常是消化性溃疡的一个重要的诱导因素[2-3]。目前,不法分子将化学药物非法加入中成药及保健食品中的现象时有发生[4-5],这些化学药物的不当应用会造成严重的不良后果,给人们的生活造成严重的威胁[6]。而胃药非法添加的检测方法尚未健全[7-10],本研究针对抗消化性溃疡类中成药及保健食品中非法添加的17种化学药物(包括胃泌素受体阻断药丙谷胺,增强胃黏膜屏障功能的药物米索前列醇,H2受体阻断药西咪替丁、雷尼替丁、法莫替丁、尼扎替丁,质子泵抑制药奥美拉唑、兰索拉唑、泮托拉唑、雷贝拉唑,M胆碱受体阻断药阿托品、东莨菪碱、山莨菪碱、溴丙胺太林,抗幽门螺杆菌药物阿莫西林、克拉霉素、甲硝唑)研究建立了采用超高效液相色谱串联三重四级杆质谱仪(UPLC-MS/MS)进行定性定量同步检测的方法。以完善对此类药物非法添加的检测,有效应对此类化学药物非法添加的监管要求。
1 仪器与试剂
LC-20A型超高效液相色谱仪(日本岛津公司)串联API3200型三重四级杆质谱仪(美国AB SCIEX公司);Shim-pack XR-ODSⅡ型色谱柱(日本岛津公司,2.0 mm×75 mm,2.2 μm);W-滤膜0.22 μm(天津市兰博实验仪器设备有限公司),VGT-2120QTD型超声清洗仪(广东固特超声股份有限公司);BP211D型电子天平(德国Sartorius公司)。
对照品丙谷胺、米索前列醇、西咪替丁、盐酸雷尼替丁、法莫替丁、尼扎替丁、奥美拉唑、兰索拉唑、泮托拉唑钠、雷贝拉唑钠、硫酸阿托品、消旋山莨菪碱、氢溴酸东莨菪碱、溴丙胺太林、甲硝唑、克拉霉素、阿莫西林批号分别为100176-201104、410004-201502、100158-201406、100163-201607、100305-201304、100853-201502、100367-201706、100709-201304、100575-201505、100658-201705、100040-201312、100249-201303、100049-201310、100471-201402、100191-201507、130558-201303、130409-201512,含量分别为99.7%、99.8%、98.9%、99.9%、99.5%、99.6%、100.0%、99.8%、95.8%、99.7%、97.2%、99.8%、99.6%、98.6%、100.0%、97.2%、86.9%,以上17种对照品均购自中国食品药品检定研究院。实测样品来自于市场抽检。乙腈、甲酸和乙酸铵均为色谱纯,购自美国Fisher试剂公司。水为超纯水。
2 方法与结果
2.1 色谱条件 岛津Shim-pack XR-ODSⅡ(2.0 mm×75 mm,2.2 μm)色谱柱,柱温20℃,流速0.25 mL/min,进样量5 μL。ESI正离子扫描模式(ESI+)流动相系统:以乙腈为流动相A,0.1%甲酸水溶液(含7 mmol/L的乙酸铵)为流动相B,梯度洗脱(0~8 min,90%~70%B;8~10 min,70%~60%B;10~13 min,60%~90%B;13~15 min,90%B),采集时间15 min;ESI负离子扫描模式(ESI-)流动相:乙腈-15 mmol/L乙酸铵(50:50),采集时间5 min。
2.2 质谱条件 离子源:电喷雾离子化源(ESI),正、负离子扫描模式;离子喷射电压(IS):5 000 V;离子源温度(TEM):500℃;源内气体GS1(Nebulizer gas,N2):25 psi;辅助加热气GS2(Heated gas,N2):45 psi;气帘气体(Cur Gas):35 psi。监测方式MRM。17种物质的准分子离子峰、主要离子碎片峰及其质谱数据采集参数见表1。

表1 17种抗溃疡病药的质谱采集参数
2.3 溶液的制备
2.3.1 对照品溶液的制备 ESI+模式对照品溶液的制备:精密称取阿莫西林、西咪替丁、法莫替丁、甲硝唑、山莨菪碱、东莨菪碱、阿托品、雷贝拉唑、奥美拉唑、泮托拉唑、克拉霉素、兰索拉唑、溴丙胺太林、丙谷胺对照品各约10 mg,分别置于20 mL棕色量瓶用50%乙腈水溶液配制成对照品储备液,冷藏。根据离子化效率的不同,精密量取溴丙胺太林储备液0.04 mL,硫酸阿托品、奥美拉唑、兰索拉唑、泮托拉唑钠、克拉霉素储备液各0.1 mL,氢溴酸东莨菪碱、消旋山莨菪碱、雷贝拉唑钠储备液各0.2 mL,西咪替丁、丙谷胺储备液各0.3 mL,甲硝唑储备液0.8 mL,法莫替丁、阿莫西林储备液各3 mL置于同一25 mL棕色量瓶中,用50%乙腈配置成ESI+混合对照品中间储备液。精密量取混合对照品中间储备液2 mL置50 mL量瓶中,用10%乙腈水溶液定容,摇匀,滤过(文中所述“滤过”均为用0.22 μm微孔滤膜滤过)。
ESI-模式对照品溶液的制备:精密称取雷尼替丁、尼扎替丁、米索前列醇对照品约10 mg,分别置于20 mL棕色量瓶用50%乙腈水溶液配置成对照品储备液,冷藏。精密量取米索前列醇对照品储备液5 mL,盐酸雷尼替丁、尼扎替丁储备液各1 mL置于同一25 mL棕色量瓶中,用50%乙腈配置成ESI-混合对照品中间储备液。精密量取混合对照品中间储备液2 mL置50 mL量瓶中,用10%乙腈水溶液定容,摇匀,滤过。
2.3.2 供试品溶液的制备 取待测样品1次服用剂量分别置于研钵中,研磨均匀成粉末状,置于50 mL量瓶中,加50%乙腈40 mL,冰浴中超声处理10 min,用50%乙腈定容,摇匀,过滤,精密量取续滤液1 mL至100 mL量瓶中,用10%乙腈定容,摇匀;再精密量取1 mL至10 mL量瓶中,用10%乙腈定容,摇匀,滤过。
2.3.3 阴性对照溶液的制备 取待测样品的阴性样品,按“2.3.2”项下方法制成空白基质粉末,分别取1次服用剂量,置于50 mL量瓶中,同法制备阴性对照溶液。
2.4 定性分析 取对照品溶液进样测定,将所得图谱中各成分定性离子对之间的相对离子丰度和色谱峰保留时间进行统计分析[11-13]。各化学药定量离子对的MRM色谱图见图1,由图1可知,保留时间相近的上述各化学药在MRM采集模式下均能达到完全分离。
取供试品溶液进样测定,记录所得质谱MRM色谱图(图2),根据所得液相质谱图,以定性离子对之间的相对丰度和保留时间定性[12],结果表明,供试品中含有西咪替丁和丙谷胺。
取“2.3.3”项下的阴性对照溶液进样测定,得到阴性对照溶液的TIC和MRM色谱图,均未见有干扰17种目标化学药成分的色谱峰。
2.5 定量分析 对判定供试品溶液中含有的化学药物,根据相应对照品定量离子对和阳性化学药物定量离子对的峰面积之比等于对照品浓度与阳性化学药物的浓度之比的关系进行含量计算[14]。
2.5.1 线性考察与检测限(LOD)和定量限(LOQ)试验 取ESI+和ESI-两种测定模式下的混合对照品中间储备液各0.05、0.1、0.2、0.5、1.0、2.0、5.0 mL,分别用10%乙腈稀释至50 mL,进样测定。以浓度为横坐标,以定量离子对色谱峰面积为纵坐标做标准曲线,得17种化学药的线性范围、线性回归方程和相关系数见表2。结果表明,17种化学药均呈良好的线性关系,相关系数均大于或等于0.995 1。
取混合对照品溶液适量,适当稀释,添加到阴性样品中,制成质量浓度约为0.5 mg/g的供试品,进样,以信噪比为3时各化学药的LOD和信噪比为10时的LOQ的计算结果见表2,表明方法的灵敏度完全能满足非法添加化学药物的检测要求。
2.5.2 精密度试验 取混合对照品溶液1份,连续进样6次,得17种化学药物的定量离子对质谱峰面积的RSD,其值均在0.9%~2.8%范围内,见表2。说明该方法的精密度良好。
2.5.3 重复性试验 取阴性样品加入适量混合对照品储备液,混匀,取此样品6份,按供试品的制备方法制备并测定,计算各成分含量的RSD值均在0.8%~3.0%,见表2。表明重复性较好。
2.5.4 稳定性试验 取2.5.3项下的同一供试品溶液,分别于制备0、2、4、6、8 h后依法测定,根据上述17种化学药的定量离子对峰面积计算RSD均在1.0%~4.6%,见表2,说明供试品溶液在测定条件下8 h内稳定。
2.5.5 加样回收试验 取一次服用量的阴性样品6份,分别置50 mL量瓶中,各加相应的混合对照品储备液适量,混匀,按供试品的制备方法制备,测定。17种化学药的平均回收率见表2。从表中可以看出,各化学药的标准加样回收率均在81.5%~102.7%。
2.5.6 样品测定 本研究收集相关中成药及保健食品共10个样品,取各样品按“2.3.2”项下方法制备供试品溶液,经测定,在10个样品中检出2个样品添加了抗消化性溃疡类化学药物,分别为西咪替丁和丙谷胺,MRM色谱图见图2。根据定量离子对的色谱峰面积,采用外标法计算样品中添加的化学药的量,样品中检出西咪替丁21.4 mg/g,丙谷胺18.1 mg/g。
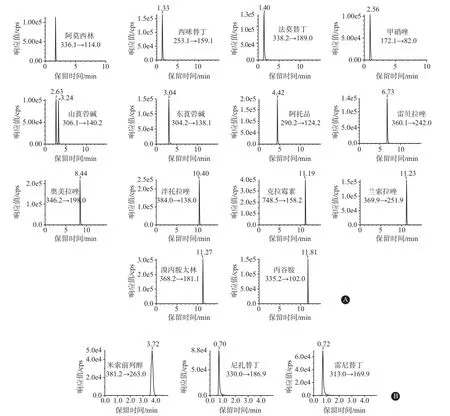
图1 17种抗溃疡病药对照品定量离子对应的MRM色谱图:A为ESI+扫描模式,B为ESI-扫描模式

图2 样品溶液中西咪替丁、丙谷胺的定性、定量离子对MRM色谱图
3 讨论
本研究分别考察了以水、不同浓度的甲酸、乙酸、乙酸铵为水相,以甲醇、乙腈为有机相,将水相和有机相组成的各种组合为流动相体系,结果发现,ESI+模式下,乙腈-0.1%甲酸(含7 mmol/L乙酸铵)流动相体系梯度洗脱,14种化合物的峰形和分离情况最好;ESI-模式下,以乙腈-15 mmol/L乙酸铵为流动相等度洗脱,3种化合物峰形和分离情况最好。在质谱条件优化过程中,发现米索前列醇在负离子模式下灵敏度高,噪声小,而其它16种化合物正离子模式灵敏度高,但是雷尼替丁和尼扎替丁在质谱ESI+模式和液相相应的流动相下,峰形很差,基线高,峰底宽度超过3 min,不利于进行定量定性,而这两种化合物在ESI-模式下,虽然灵敏度相对于ESI+稍低,但在相应的流动相下,基线和峰形好,适用于定量和定性,故选择ESI-模式检测米索前列醇、雷尼替丁和尼扎替丁,用ESI+模式检测其它14种化合物。
因质子泵抑制药不耐酸,因此口服质子泵抑制剂药大多为肠溶包衣制剂,以避免药物因胃酸作用而造成生物利用度降低[15];米索前列醇的纯品化学性质不稳定,需存放于-20℃环境中,且在酸性和碱性条件下都能发生化学反应,而它却可以在常温下稳定的保存于羟丙基甲基纤维素的分散体系中[16],故米索前列醇被分散于此介质中制成药剂使用。基于上述原因,将质子泵抑制药和米索前列醇的原料药作为非法添加物可能性很小,但是在日常检验工作中发现,不法分子将化学制剂成品甚至过期药添加入中成药和保健品中的现象时有发生,因而不排除质子泵抑制剂和米索前列醇以稳定形式被非法添加的可能性。
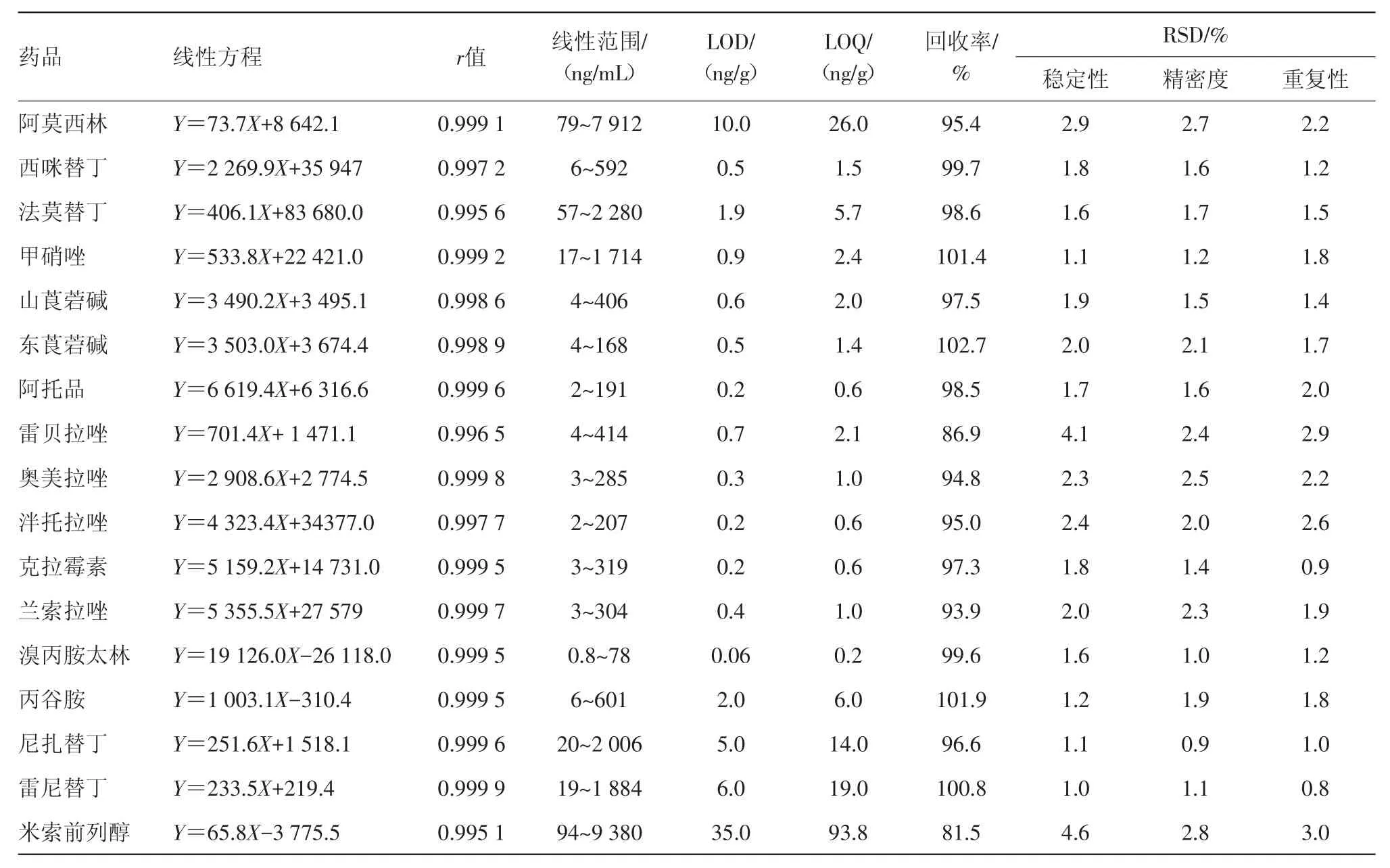
表2 17种抗溃疡病药的线性方程、相关系数、回收率及检出限、定量限
