联合有氧-抗阻运动对2型糖尿病小鼠骨髓内皮祖细胞血管生成功能的影响及其可能机制▲
2020-01-02黄燕凤刘玉花
黄燕凤 翟 露 刘玉花 马 翠 戴 霞
(广西医科大学第一附属医院护理部,南宁市 530021,电子邮箱:1319376554@qq.com)
糖尿病是以血糖升高为特征的代谢性疾病,其可引起血管内皮损伤和功能异常,促进动脉粥样硬化的发生发展,导致冠心病、缺血性脑卒中及下肢动脉闭塞等的发生,并发症的发生是糖尿病患者致死致残的主要原因[1-2]。而这种血管内皮功能障碍的发生与内皮祖细胞(endothelial progenitor cells,EPCs)密切相关[3]。EPCs是血管内皮细胞的前体细胞,在生理或病理因素刺激下,EPCs可从骨髓向靶部位动员、归巢及分化,参与血管新生及损伤血管的再内皮化[4]。然而,糖尿病发生时机体EPCs数量减少、功能受损,不能有效地发挥内皮修复作用[5-6]。因此,探寻改善糖尿病状态下EPCs血管生成功能的方法及其相关机制,对治疗糖尿病血管病变有重要意义。
有研究表明,有氧运动能提高中老年大鼠的EPCs增殖功能,促进损伤血管内皮的修复[7]。但目前关于运动影响糖尿病患者或者动物模型EPCs功能作用的研究较少,尚未发现有研究报告联合抗阻-有氧运动(简称联合运动)对2型糖尿病患者EPCs功能的影响,其相关机制尚不十分明确。有学者报告,磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)信号通路参与血管修复和生成过程,激活PI3K/AKT信号通路可以改善糖尿病小鼠EPCs血管生成能力[8]。联合运动是否能通过调节PI3K/AKT的表达水平,进而改善EPCs的血管生成功能有待进一步研究。本研究对2型糖尿病小鼠实施为期8周的联合运动干预,体外分离培养骨髓源EPCs,探讨联合运动对EPCs血管再生功能的影响及其相关机制,为运动治疗糖尿病、防治糖尿病血管并发症提供参考依据。
1 材料和方法
1.1 动物及分组 选择20只2型糖尿病db/db小鼠[品系名称:BKS-DB/Nju;购自南京大学-南京生物医药研究院,许可证号:SYXK(苏)2016-0012],8周龄,均为雄性,平均血糖为28 mmol/L,平均体重为42 g。按照随机数字表法,将小鼠分为联合运动组和空白组,每组10只。
1.2 运动训练 空白组小鼠不进行任何运动干预。联合运动组采取有氧运动与抗阻运动相交替的训练方式,每周运动6 d,即周一、三、五进行抗阻运动,周二、四、六进行有氧运动,共8周。其中,抗阻运动为在小鼠的尾部缠绕一定重量的水球促其进行爬梯训练,具体干预方案见表1;有氧运动为小鼠在坡度均为0度的8跑道小鼠跑台(北京东西仪器科技有限公司,型号:WDW-1)上跑步,第1周时有氧运动速度为10 m/min,30 min/d,逐渐增加至15 m/min,30 min/d,第2~8周速度为15 m/min,60 min/d。

表1 抗阻运动方案
注:BW为自身体重。
1.3 主要试剂及仪器 淋巴细胞分离液(天津市灏洋生物制品科技有限责任公司,批号:LTS1092),EGM-2MV培养基(Lonza公司,批号:CC-3162),纤维粘连蛋白(RD公司,批号:1918-FN-02M),T25细胞培养瓶(Corning公司,批号:430168),DiI标记的乙酰化低密度脂蛋白(DiI-labelled acetylated low density lipoprotein,DiI-ac-LDL;Molecular Probes公司,批号:L3484),异硫氰酸荧光素标记的荆豆凝集素Ⅰ(fluorescein isothiocyanate-labelled ulex europaeus agglutinin Ⅰ,FITC-UEA-Ⅰ;Sigma公司,批号:L9006),PI3K/AKT通路特异性抑制剂LY294002(MedChemExpress公司,批号:B524631337769),BD MatrigelTM基底膜基质(Corning公司,批号:354230),μ-Slide Angiogenesis板(ibidi公司,批号:81506),cDNA合成试剂盒(Thermo Fisher Scientific公司,批号:K1622),实时荧光定量PCR试剂盒(QIAGEN公司,批号:208054),放射免疫沉淀(radio immunoprecipitation assay,RIPA)裂解液(碧云天公司,批号:P0013B),苯甲基磺酰氟溶液(索莱宝公司,批号:P8340),蛋白磷酸酶抑制剂(索莱宝公司,批号:P1260)。一抗:抗β-肌动蛋白单克隆抗体(索莱宝公司,批号:K200058M)、AKT抗体(CST公司,批号:9272s)、PI3Kp85(19H8)兔单克隆抗体(CST公司,批号:4257s);二抗:兔抗小鼠IgG(碧云天公司,批号:P0023D)。激光共聚焦显微镜(日本Olympus公司,型号:OLYMPUS CKX53)。
1.4 EPCs体外分离培养及鉴定 运动干预第8周结束后处死小鼠进行取材。无菌分离小鼠肱骨、股骨及胫骨并离断,以含双抗的无菌磷酸缓冲盐溶液(phosphate-buffered saline,PBS)充分冲洗骨髓腔,冲洗时间至少5 min,将收集的骨髓腔冲洗液轻柔吹打均匀后,按1 ∶1体积比缓慢滴加于淋巴细胞分离液上,4℃下2 000 r/min离心20 min后用巴氏吸管吸取出中间白色云雾状细胞层,加入PBS清洗细胞以去除残留的淋巴细胞分离液,4℃下2 000 r/min离心5 min获取细胞沉淀,加入EGM-2MV培养基重悬细胞。将细胞培养物接种于包被了纤维粘连蛋白的T25细胞培养瓶中,放置于5% CO2培养箱中,37℃恒温培养。每天观察细胞生长情况,每3天换液1次,去除非贴壁细胞。待细胞汇合度达到80%,进行细胞传代培养并进行EPCs的鉴定。取EPCs以2.5×105个/孔的密度平铺于24孔培养板,加入1 mL DiI-ac-LDL,使Dil-ac-LDL最终浓度为10 μg/mL,5% CO2、95%饱和湿度培养箱孵育4~6 h,1×PBS缓冲液冲洗3次;再取1 mL FITC-UEA-Ⅰ加入24孔板中,使FITC-UEA-Ⅰ最终浓度10 μg/mL,5% CO2、95%饱和湿度培养箱孵育1~2 h后,1×PBS缓冲液冲洗3次。使用激光共聚焦显微镜进行观察,Dil-ac-LDL、FITC-UEA-Ⅰ双染色阳性的细胞被认为是正在分化的EPCs。随机选取10个视野,进行细胞计数并计算EPCs双阳性率。
1.5 PI3K/AKT特异性抑制剂处理细胞 将联合运动组细胞分为联合运动组和联合运动+抑制剂组,对照组细胞分为对照组和对照+抑制剂组。将EPCs接种于12孔板,加入1 μL、浓度为0.7 μmol/L的LY294002至各抑制剂组,于37℃培养箱中孵育1 h,联合运动组与对照组不给予任何处理。
1.6 EPCs体外血管生成功能实验 用BD MatrigelTM基底膜基质包被μ-Slide Angiogenesis板,铺板完成后,将其置于4℃冰箱平衡30 min后于37℃培养箱中孵育1 h使BD MatrigelTM基底膜基质凝固。取1.5干预后的EPCs,加入0.5 mL的0.25%胰蛋白酶消化细胞,使其脱壁后常温下以1 000 r/min离心3 min,弃上清,以完全培养基重悬细胞,将细胞密度调整至5×104个/mL,分别取100 μL加入BD MatrigelTM基底膜基质包被的μ-Slide Angiogenesis板中,放入37℃、5% CO2培养箱培养24 h后取出培养板,并进行拍照。采用Image J图像分析软件对形成的完整管腔进行计数(每3个分叉处记为1个血管腔)。以体外新生血管的主干长度和节点作为评价体外血管生成功能的标准。
1.7 实时荧光定量PCR检测PI3K、AKT mRNA的表达 用TRIzol法提取各组细胞的总RNA并检测RNA纯度,按cDNA合成试剂盒操作说明步骤进行扩增,设置反应条件:42℃ 60 min,70℃ 5 min。反转录后,取适量反转录产物按实时荧光定量PCR试剂盒操作说明进行扩增,反应体系共10 μL,包括2×绿色荧光实时聚合酶链反应混合液5 μL、荧光染料0.05 μL、终浓度为0.5 μmmol/L的上下游引物各0.5 μL、cDNA 1 μL,加无RNA酶水至10 μL;设置扩增反应条件为95℃预变性2 min后,95℃ 5 s,60℃ 30 s扩增40个循环。各样品均重复3次,采用2-ΔΔCT法计算相对表达水平,以β-肌动蛋白作为内参,各基因引物见表2。

表2 基因引物序列
1.8 Western blot检测AKT、p-AKT蛋白的表达 取各组细胞置于25 cm2细胞培养瓶中,加入RIPA裂解液、苯甲基磺酰氟溶液、蛋白磷酸酶抑制剂混合液以裂解EPCs,使用二喹啉甲酸法测定蛋白浓度,将蛋白进行10%十二烷基硫酸钠-聚丙烯酰胺凝胶电泳分离并转到聚偏二氟乙烯膜上。将聚偏二氟乙烯膜置于含有5%脱脂奶粉的玻璃皿上封闭1 h后,1×洗涤缓冲液(tris buffered saline/tween,TBST)溶液洗涤3次,加入一抗室温摇床孵育2 h,1×TBST溶液洗涤3次(20 min/次)后加入二抗进行室温摇床孵育1 h,最后用Odyssey双色红外激光成像系统(美国LI-COR公司)扫描,选择800通道,调节明亮度,记录各组蛋白灰度值。
1.9 统计学分析 采用SPSS 17.0软件进行统计学分析。计量资料以(x±s)表示,组间比较采用单因素方差分析,两两比较采用Tukey法。以P<0.05为差异有统计学意义。
2 结 果
2.1 EPCs形态 骨髓分离后获得的EPCs在纤维粘连蛋白包被的培养瓶中培养,4 d后EPCs大多呈圆形、短梭形,部分为长梭形;7 d后圆形细胞数量减少,梭形细胞数量渐渐增多,可出现细胞集落;10 d后EPCs大量增殖,并有梭形贴壁细胞在细胞集落边缘不断生成,可出现“铺路石”样改变。见图1。

图1 倒置显微镜下EPCs的形态(×100)
注:A为体外培养第48 h后,部分细胞开始贴壁,少量细胞呈梭形;B为7 d后,EPCs处于贴壁状态,多呈梭形或者类圆形;C为12 d后,梭形细胞增多,逐渐长满培养瓶底部,可见有“铺路石”状改变。
2.2 EPCs的鉴定结果 在激光共聚焦显微镜下可观察到,胞浆摄取DiI-ac-LDL,显示红色,胞膜结合FITC-UEA-Ⅰ,显示绿色,双染色阳性细胞为黄色,为正在分化的EPCs。DiI-ac-LDL及FITC-UEA-Ⅰ双染阳性率为(73.50±7.47)%。见图2。
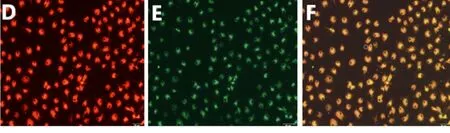
图2 DiI-ac-LDL及FITC-UEA-Ⅰ双染试验结果(×200)
注: DiI-ac-LDL染色阳性细胞发红色荧光(D),FITC-UEA-Ⅰ染色阳性细胞发绿色荧光(E),两者双染阳性细胞呈黄色荧光(F)。
2.3 各组EPCs的体外血管生成功能 对照组与对照+抑制剂组EPCs的主干长度和节点比较,差异无统计学意义(P>0.05),而联合运动组EPCs的主干长度和节点均长于或多于对照组及联合运动+抑制剂组(均P<0.05),见图3和表3。

图3 各组EPCs体外血管生成功能情况(×100)
注:H为对照组,I为对照+抑制剂组,J为联合运动组,K为联合运动+抑制剂组。
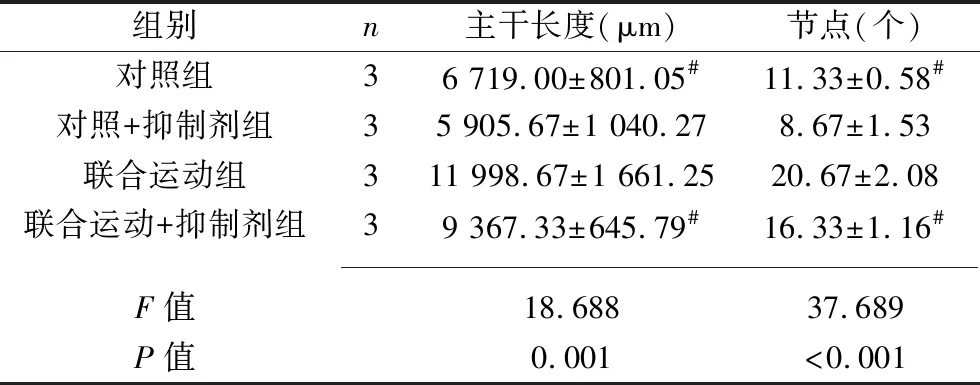
表3 各组EPCs的血管生成主干长度和节点比较(x±s)
注:与联合运动组比较,#P<0.05。
2.4 各组EPCs的PI3K、AKT mRNA表达情况 与对照组比较,对照+抑制剂组的PI3K、AKT mRNA相对表达水平均降低,而联合运动组的PI3K、AKT mRNA相对表达水平均升高(均P<0.05);联合运动+抑制剂组的PI3K、AKT mRNA相对表达水平均低于联合运动组(均P<0.05)。见表4。

表4 各组细胞AKT和PI3K的mRNA相对表达水平比较(x±s)
注:与对照组比较,*P<0.05;与联合运动组比较,#P<0.05。
2.5 各组EPCs的AKT、p-AKT蛋白表达情况 与对照组比较,对照+抑制剂组AKT蛋白的相对表达水平降低,联合运动组的AKT、p-AKT蛋白的相对表达水平均升高(均P<0.05);联合运动+抑制剂组AKT、p-AKT蛋白的相对表达水平均低于联合运动组(均P<0.05)。见表5。
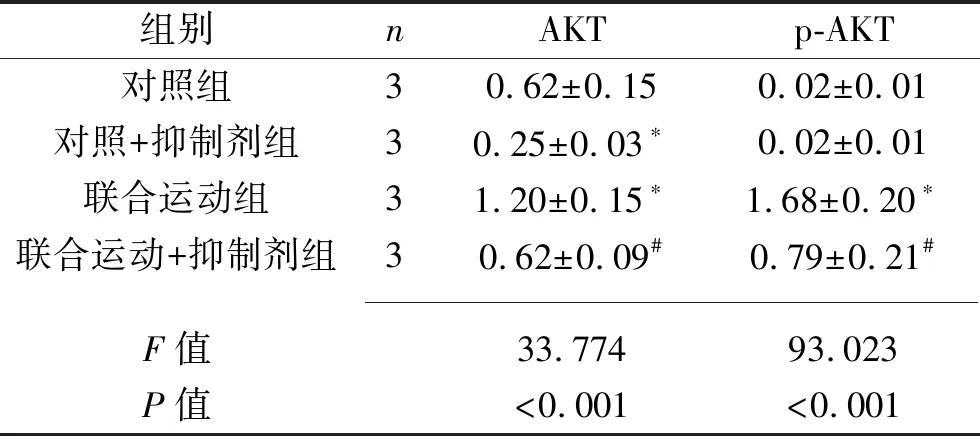
表5 各组EPCs的AKT和p-AKT蛋白相对表达水平比较(x±s)
注:与对照组比较,*P<0.05;与联合运动组比较,#P<0.05。
3 讨 论
研究表明,当血管内皮受损时,EPCs可以从骨髓动员,并迁移、整合到血管网络,通过分化为成熟的内皮细胞从而修复损伤血管和生成新的血管[4]。而2型糖尿病会导致EPCs血管生成功能减弱,影响损伤内皮的修复[9]。有文献报告,联合运动较单纯有氧或抗阻运动能更有效地改善糖尿病前期人群的糖代谢[10]。然而,目前联合抗阻-有氧运动对2型糖尿病患者EPCs血管生成功能的作用尚未十分明确,相关机制也尚未清楚。本研究结果显示,联合运动组EPCs的主干长度和节点均长于或多于对照组(P<0.05),即联合运动组呈现更强的血管修复能力,提示联合运动训练可增强骨髓EPCs的体外血管生成功能。
为了明确联合运动改善EPCs功能活性的可能相关机制,本研究对EPCs上参与血管修复和生成的PI3K/AKT通路进一步探究。PI3K的活化很大程度上依赖于靠近其质膜内侧的底物,即多数生长因子和信号传导复合物,包括成纤维细胞生长因子、血管内皮生长因子、人生长因子等,都能启始PI3K的激活过程[11]。AKT是一类蛋白激酶家族的丝氨酸/苏氨酸激酶,当组织损伤后,受损部位能够释放某些细胞因子如血管内皮生长因子,从而激活PI3K/AKT信号通路[12]。有研究表明,PI3K/AKT的激活与EPCs的体外血管生成功能密切有关,参与血管损伤修复过程[8]。Dai等[13]发现阻断PI3K/AKT的磷酸化激活,可抑制下肢缺血模型糖尿病小鼠的EPCs体外血管生成能力,减少新生血管长度。而其他学者发现,为期14天的游泳干预可促进小鼠EPCs从骨髓动员到外周血,增加了外周血EPCs的数量,新生血管密度升高,并上调PI3K和AKT的表达水平;而使用PI3K/AKT通路抑制剂,可抑制小鼠的EPCs动员和血管生成功能[14]。本研究结果显示,与对照组比较,联合运动组AKT、p-AKT蛋白相对表达水平上调(P<0.05),EPCs血管生成主干长度和节点较对照组增加,这表明联合运动可通过上调AKT,改善2型糖尿病小鼠EPCs血管生成功能。本研究进一步使用PI3K/AKT抑制剂进行干预观察,结果显示,与对照组比较,对照+抑制剂组的PI3K、AKT的 mRNA下调(P<0.05),AKT、p-AKT蛋白相对表达量下降(P<0.05),而两者EPCs的主干长度和节点差异无统计学意义(P<0.05),即PI3K/AKT抑制剂可有效抑制2型糖尿病小鼠EPCs AKT蛋白的表达,但对其血管生成功能无明显影响。与未加入PI3K/AKT抑制剂的联合运动组相比,联合运动+抑制剂组PI3K、AKT的 mRNA下调(P<0.05),AKT、p-AKT蛋白相对表达量下降(P<0.05),同时EPCs主干长度和节点减少(P<0.05),即抑制AKT的表达水平,可削弱联合运动对2型糖尿病小鼠EPCs体外血管生成能力的干预效果。
总之,8周的联合运动能有效改善2型糖尿病小鼠EPCs的血管生成能力,其机制可能与上调AKT密切相关,抑制AKT的表达可削弱其干预效果。本研究为运动防治糖尿病血管并发症提供新的基础研究依据和干预靶点,有关联合运动调节EPCs改善血管内皮损伤的具体机制还有待进一步研究。
