联合应用RNA-seq和ATAC-seq寻找FOXQ1转录因子的下游靶基因
2020-01-02伊梦杰张锦锦
伊梦杰,张锦锦,郭 强,唐 慧*
(1.昆明理工大学 医学院, 昆明 650504;2.云南省第一人民医院,昆明理工大学附属医院,云南省消化内镜临床医学中心,云南省临床病毒学重点实验室,昆明市肿瘤分子与免疫防治重点实验室,昆明 650032;3.中国科学院昆明动物研究所,昆明 650223)
结直肠癌(Colorectal cancer, CRC)是一种由多致病因素导致且预后很差的恶性消化道肿瘤,是世界第三大常见肿瘤,其死亡率在所有癌症中居于第四位(位于肺癌、肝癌、胃癌之后)[1]。叉头框Q1(Forkhead box Q1,FOXQ1)是叉头框(Forkhead box,FOX)基因家族的成员之一,基因定位于6p23-25,编码含403个氨基酸的FOXQl蛋白,作为一类核转录因子,可以稳定结合到靶基因启动子区域的GC盒等核心元件,控制下游基因转录活性从而发挥生物效应[2]。2010 年Kaneda等[3]研究发现FOXQ1在CRC中异常高表达,近年来大量研究亦证实FOXQ1在卵巢癌、乳腺癌、膀胱移形细胞癌、胃癌、肝癌、非小细胞肺癌和神经脑胶质瘤等多种肿瘤中异常表达[4-6],与多种肿瘤的发生、发展密切相关,在许多肿瘤中具有明确的促肿瘤生长转移的功能。因此,鉴定新的FOXQ1下游靶基因,丰富由FOXQ1参与并介导的信号通路信息,有望为肿瘤的靶向治疗提供新的靶点。
ATAC-seq是一种检测染色质易接近性的测序方法,可以检测样本间染色质易接近性的变化情况。ATAC-seq只需要很少的细胞(50 000个)就能检测基因组中所有活跃的调控序列[7],DNA探针(作为转座子发挥作用)通过酶促反应(转座酶Tn5)被整合到基因组的开放区域,然后通过测序来鉴定这些区域[8]。本实验联合应用RNA-seq与ATAC-seq寻找由FOXQ1敲低引起的染色质易接近性改变所导致的表达改变基因,即FOXQ1转录因子调控的潜在下游基因。
1 材料和方法
1.1 细胞、仪器和试剂
本实验所用细胞系均购于中科院上海细胞库,DMEM高糖培养液、RPMI 1640培养液购于美国Corning公司,胎牛血清购于美国Gibco公司。Puromycin购于北京索莱宝科技有限公司,FOXQ1一抗购于abcam公司,HRP-Rb-anti-goat二抗购于Cell Signaling Technology公司,β-actin一抗、HRP-goat-anti-mouse二抗均购于Proteintech公司。RNAzol RT RNA Isolation Reagent购于美国MRC公司,引物由宝生物工程有限公司合成。核酸定量仪为Thermo公司产品,LightCycler 480实时荧光定量仪为Roche公司产品,WB垂直基础电泳仪为伯乐公司产品。
1.2 方法
1.2.1 实验分组
根据课题组前期工作并结合文献报道,选取在CRC细胞系中FOXQ1表达量较高的SW480和DLD1细胞系进行FOXQ1基因的敲低实验,实验分为两组共4株细胞。FOXQ1基因敲低组:SW480-shFOXQ1和DLD1-shFOXQ1;对照组:SW480-shControl和DLD1-shControl。
1.2.2 FOXQ1基因低表达稳定细胞系的构建
1.2.2.1 慢病毒表达质粒的构建、筛选与扩增
根据FOXQ1序列,得到siRNA靶序列,设计合成3对shRNA干扰序列(见表1),命名为shRNA-A、shRNA-B、shRNA-C,并设计一条对照序列shRNA-control;应用Addgene的pSPAX2慢病毒包装系统,PLKO.1-puro-shFOXQ1质粒包装构建获得lenti-shFOXQ1慢病毒,每组重组质粒经测序鉴定无误后,用去内毒素大提试剂盒提取质粒,测浓度;酶切鉴定无误后,在HEK293T细胞中扩增,之后用LB培养基筛选、扩增[9]。
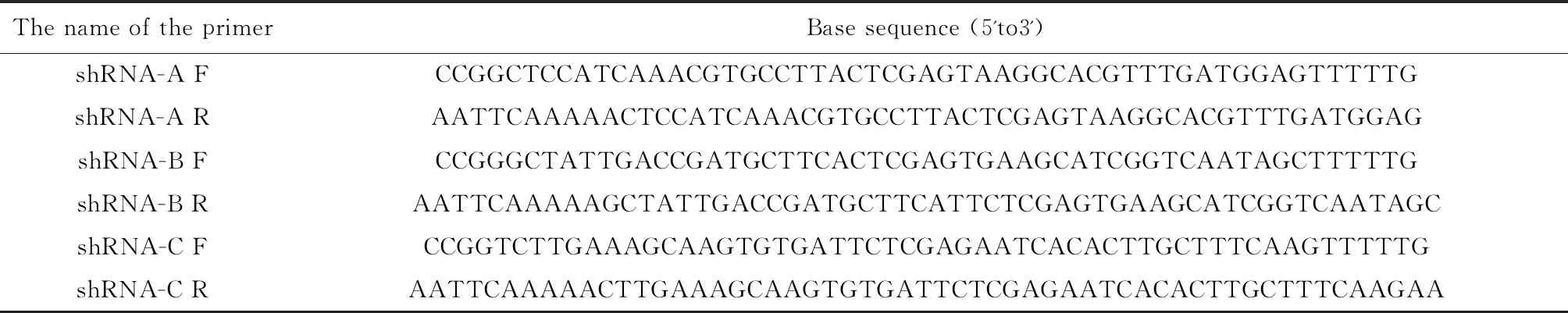
表1 shRNA引物序列Table 1 shRNA primer sequences
1.2.2.2 慢病毒的感染及阳性细胞的筛选
将待感染的SW480和DLD1细胞铺在24孔板中培养,生长密度达70%~80%时将病毒浓缩液加至细胞中,培养12~15 h后更换为完全培养基。待细胞长满时,将24孔板中的细胞传代至六孔板中进行初步扩大培养,并加入含有1 μg/ml嘌呤霉素的培养基筛选阳性细胞。筛选获得的阳性细胞分别应用qRT-PCR和WB在mRNA和蛋白水平验证FOXQ1基因的敲低效率[10]。
1.2.3 RNA-seq
1.2.3.1 收集细胞
将稳定转染的细胞传代培养稳定生长后,分别收集生长状态良好的DLD1、SW480基因敲低组与对照组细胞进行后续RNA-seq实验。
1.2.3.2 RNA的提取和检测
提取基因敲低组与对照组细胞总RNA,用Nanodrop 2000检测所提总RNA的浓度和纯度后用琼脂糖凝胶电泳检测RNA的完整性,最后利用Agilent 2100测定RIN值。
1.2.3.3 RNA的富集和测序
使用结合有poly-T寡核苷酸的磁珠从总RNA中分离出含有poly-A的mRNA,加入片段化缓冲剂将其打成片段,将片段化的mRNA逆转录成cDNA并纯化;经末端修复、poly-A添加、测序接头连接及AMPure XP beads 筛选后得出大小合适的片段,进行 PCR 扩增,建立测序文库并进行文库质控[11]。
1.2.3.4 上机测试。
对构建合格的测序文库进行双末端(Paired-end)测序,本测序实验组和对照组均设置了三次生物学重复,测序工作由上海嘉因生物科技有限公司完成。
1.2.3.5 RNA-seq的数据分析
RNA-seq数据下机后首先进行空载去除、接头去除等数据预处理后产出原始数据,紧接着利用FastQC软件进行数据质量控制,利用生物信息学软件STAR、HTSeq和DESeq2对测序结果进行参考序列比对、表达量统计、差异基因筛选等分析[12-14],并通过R软件绘制基因聚类分析图、火山图等。分别建立DLD1和SW480细胞FOXQ1敲低前后的转录谱。
1.2.4 ATAC-seq
1.2.4.1 收集细胞
分别收集DLD1、SW480基因敲低组和对照组细胞,计数50 000个细胞,离心去上清后依次用预冷的PBS、lysis buffer悬浮细胞500g,4 °C离心去除上清液,立即进行转座反应[15]。
1.2.4.2 转座反应与纯化
确保细胞始终置于冰上,配置转座反应体系悬浮细胞,37 °C孵育30 min,立即用Qiagen MinElute PCR Purification Kit纯化DNA,之后用10μl elution buffer洗脱。
1.2.4.3 PCR扩增
配置PCR反应体系循环扩增,之后用Qiagen MinElute PCR Purification Kit纯化DNA[8]。
1.2.4.4 上机测试
库检合格后,把不同文库按照有效浓度及目标下机数据量的需求合并后进行Illumina HiSeq测序。测序基于边合成边测序(Sequencing by Synthesis)的原理进行,在序的流动池中加入四种荧光标记的dNTP、DNA聚合酶以及接头引物进行扩增,在每一个测序簇延伸互补链时,每加入一个被荧光标记的dNTP就能释放出相对应的荧光,测序仪通过捕获荧光信号,并通过计算机软件将光信号转化为测序峰,从而获得待测片段的序列信息。本测序实验由上海嘉因生物科技有限公司完成。
1.2.4.5 ATAC-seq的数据分析
首先对ATAC-seq下机数据进行预处理,如FastQC质量控制,去除duplicate sequences,去除blacklist reads[16],原始序列比对,比对后去除重复序列和细胞器序列等,再利用生物信息学软件BWA、Macs2、Homer、Deep tools等对ATAC-seq测序结果进行参考序列比对分析、差异结合位点检测、特征峰在全基因组上的分布注释等,建立DLD1和SW480细胞FOXQ1敲低前后的染色质易接近性变化图谱。
1.2.5 RNA-seq和ATAC-seq数据的关联分析
分别将DLD1和SW480两个细胞系中RNA-seq的差异基因和ATAC-seq的差异表达峰进行关联分析,得到两组关联分析结果,以明确两组细胞中染色质易接近性变化区域对下游基因的调控功能,并找出每种细胞实验组与对照组相比差异染色质易接近性区域可能调控的下游基因。
1.2.6 两个细胞系关联分析数据的重叠分析
从DLD1和SW480的关联分析结果中找出两细胞共有的重叠部分,即获得FOXQ1敲低后引起的染色质易接近性改变所导致的表达改变基因。
1.2.7 代谢通路分析
将重叠分析结果中所获基因基于代谢通路数据库(Kyoto Encyclopedia of Genes and Genomes, KEGG)进行代谢通路注释,得到注释基因参与的所有代谢通路名称,采用Fisher检验计算代谢通路的显著性水平(P<0.05),从而筛选出注释基因富集的显著性代谢通路[17]。
2 结果分析
2.1 在mRNA和蛋白水平验证 DLD1和SW480细胞中FOXQ1的敲低效率
qRT-PCR和WB实验结果(见图1)表明成功构建了FOXQ1基因敲低组DLD1-sh-FOXQ1、SW480-sh-FOXQ1 以及对照组DLD1-sh-Control和SW480-sh-Control。FOXQ1基因敲低组在mRNA和蛋白水平与对照组相比FOXQ1基因的表达量均显著降低(***P<0.001)。
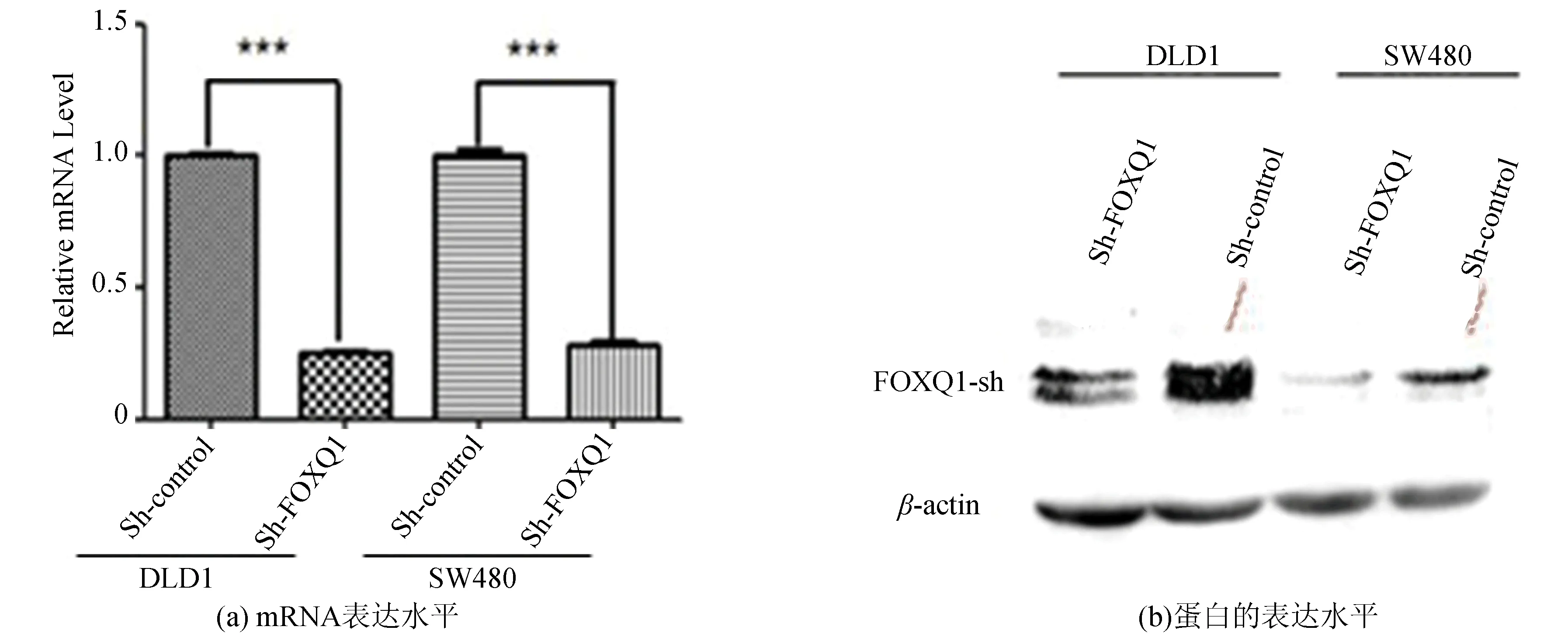
图1 稳定低表达FOXQ1的DLD1和SW480细胞验证(***P<0.001)Fig.1 Verification of the FOXQ1 expression quantity in DLD1 and SW480 cells which knock down FOXQ1 gene steadily (***P<0.001)
2.2 RNA-seq
RNA-seq结果表明,DLD1细胞FOXQ1基因敲低组与对照组相比表达显著上调的基因有215个,表达显著下调的基因有131个;SW480细胞FOXQ1敲低组与对照组相比表达显著上调的基因有171个,表达显著下调的基因有358个(表达差异基因的筛选阈值为FDR<0.05,log2FC>1)。对这些表达显著差异基因进行分析,发现在FOXQ1基因敲低后,与侵袭、自噬相关的EI24基因在表达下调基因中位居前列,与先天免疫应答和炎症反应相关的TLR2基因以及与迁移相关的SMAD3基因在表达上调的基因中位居前列。推测这些基因的表达改变与CRC的发生、侵袭、发展等密切相关,将进一步结合ATAC-seq分析这几种基因在FOXQ1敲低后,染色质开放区域的变化情况(见图2)。
2.3 ATAC-seq
ATAC-seq结果表明DLD1-shFOXQ1组有39 146个特征峰,DLD1-shControl组有49 381个特征峰,其中,基因敲低组与对照组相比染色质易接近性增强的差异表达峰有2 385个,染色质易接近性减弱的差异表达峰有6 205个。SW480-shFOXQ1组有38 962个特征峰,SW480-shControl组有42 244个特征峰,其中,基因敲低组与对照组相比染色质易接近性增强的差异表达峰有4 563个,染色质易接近性减弱的差异表达峰有3 733个(见图3)。
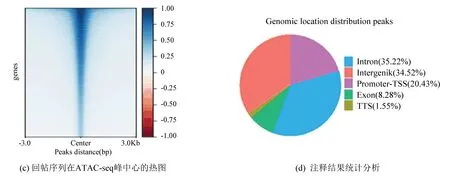
图3 ATAC-seq分析图Fig.3 ATAC-seq analysis
2.4 RNA-seq与ATAC-seq的关联分析
本实验将RNA-seq结果中的差异基因和ATAC-seq结果中差异染色质开放区域进行关联分析,预测由FOXQ1基因敲低所导致的染色质开放区域改变引起的转录因子的结合能力的改变,及最终导致下游基因的表达上调或下调。结果显示,DLD1染色质易接近性减弱区域对下游基因表达具有抑制作用(见图4(a));染色质易接近性增强区域对下游基因表达具有促进作用(见图4(b))。在SW480细胞中也观察到相同的结果(见图4(c)、图4(d))[4]。
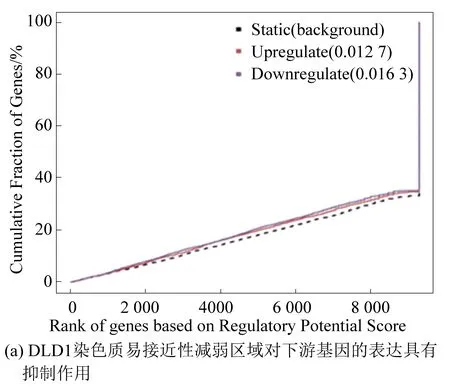

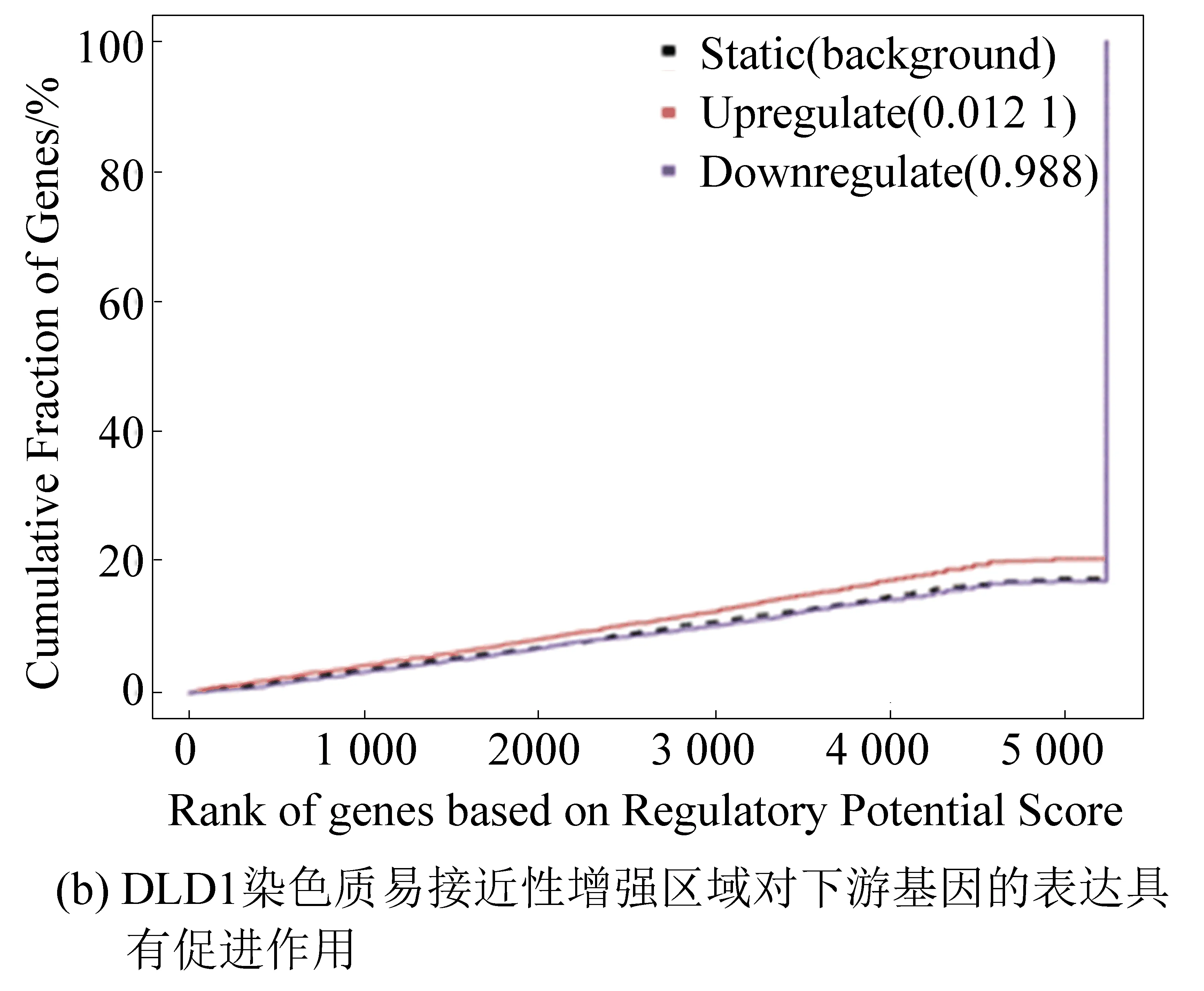
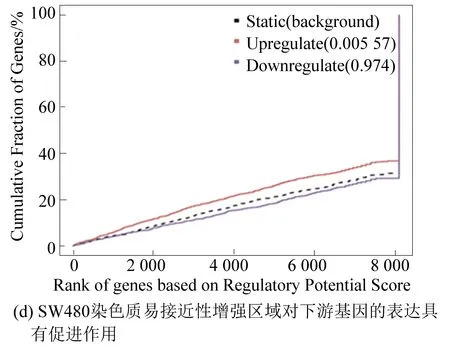
图4 染色质功能预测Fig.4 Prediction of chromatin function
2.5 SW480与DLD1关联分析结果的重叠分析
将两个细胞系的关联分析结果基因进行重叠分析。重叠部分为在两种细胞中染色质易接近性变化相同且调控的表达量变化趋势相同的基因。其中, DLD1细胞系中染色质易接近性减弱区域调控的下调基因有1 436个,SW480细胞系中染色质易接近性减弱区域调控的下调基因有407个(见图5),两细胞系染色质易接近性减弱区域共同调控的表达下调基因有70个。DLD1细胞系中染色质易接近性增强区域调控的上调基因有792个,SW480细胞系中染色质易接近性增强区域调控的上调基因有531个(见图5),两细胞系染色质易接近性增强区域共同调控的表达上调基因有61个。
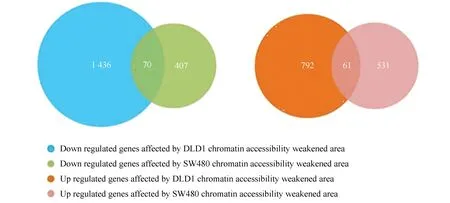
图5 SW480与DLD1关联分析结果的重叠分析Fig.5 Overlapping analysis of correlation analysis results in SW480 and DLD1
在这两个细胞的交集基因中发现EI24、TLR2、SMAD3基因也在其中,所以观察这些基因在染色质易接近性方面发生的变化(见图6~图8)。其中,TLR2、SMAD3基因在ATAC-seq数据中基因敲低组与对照组相比染色质区域有较为明显的变化,而EI24基因的染色质区域变化很弱。
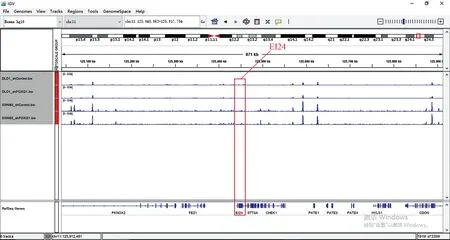
图6 ATAC-seq EI24基因比对Fig.6 ATAC-seq EI24 genome comparison
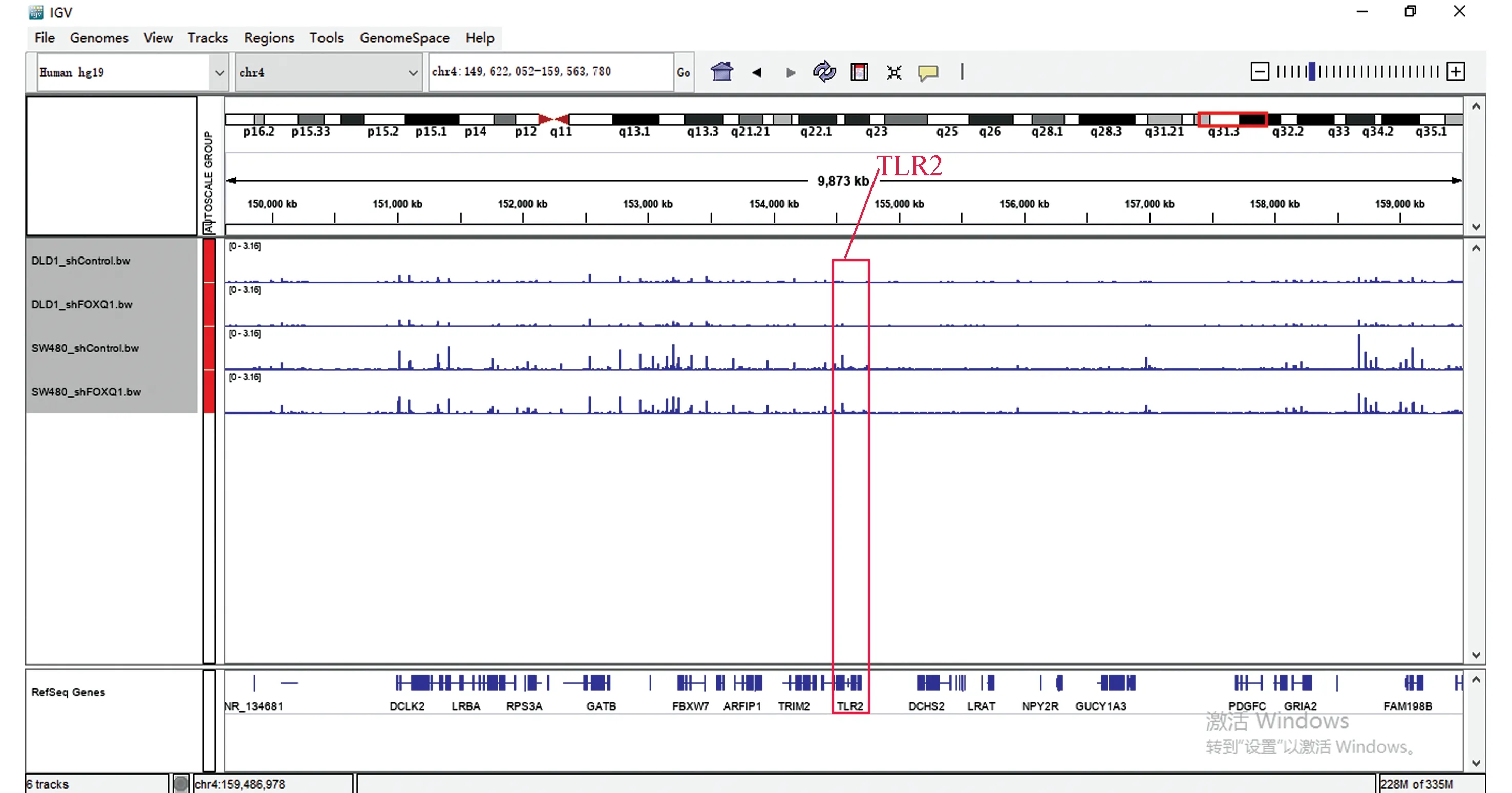
图7 ATAC-seq TLR2基因比对Fig.7 ATAC-seq TLR2 genome comparison
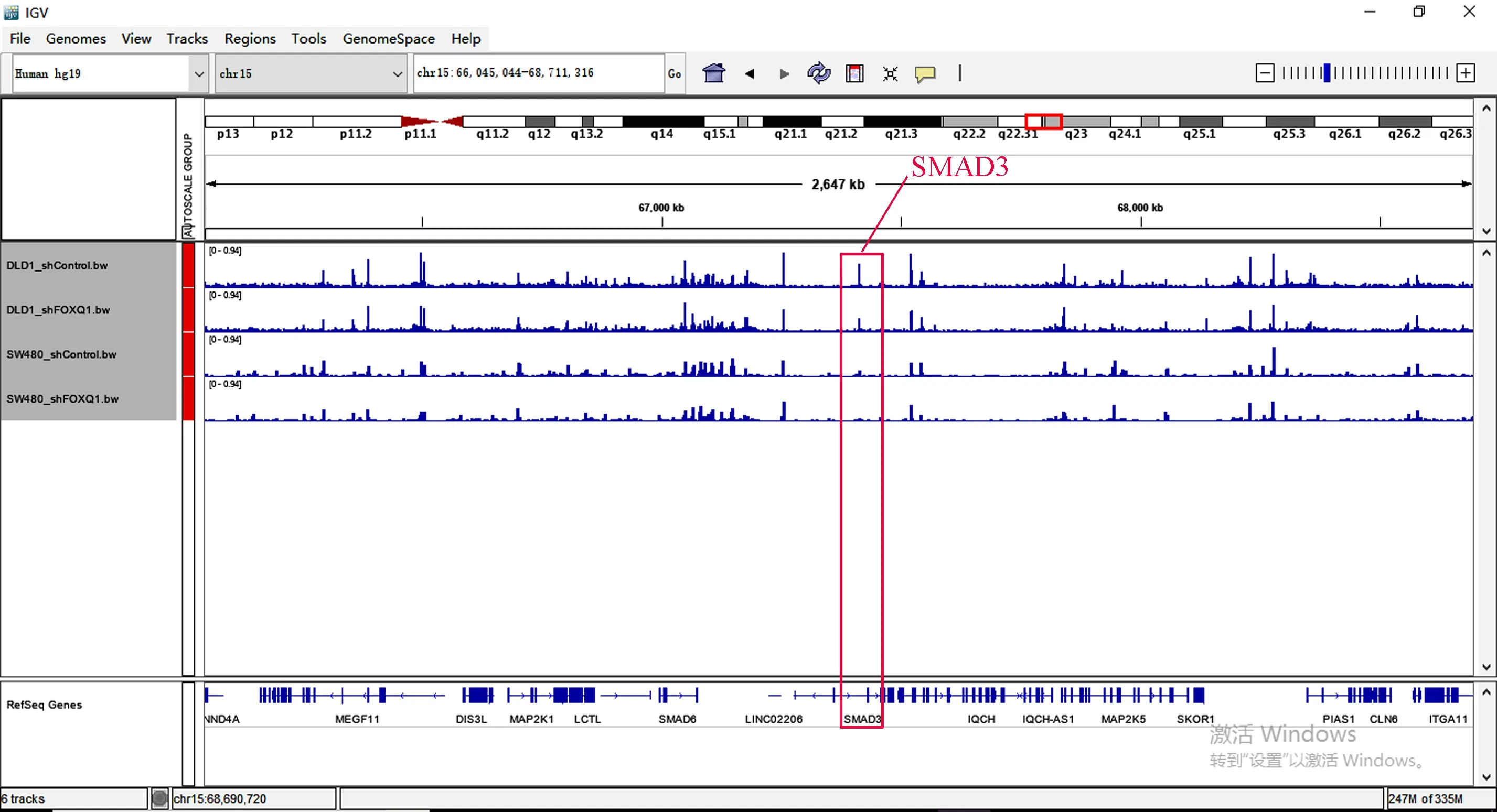
图8 ATAC-seq SMAD3基因比对Fig.8 ATAC-seq SMAD3 genome comparison
2.6 代谢通路分析
将两个细胞系的重叠基因进行代谢通路分析,结果表明染色质易接近性增强区域调控的表达上调基因显著性富集的代谢通路包括炎症性肠病(Inflammatory bowel disease, IBD)、甘油磷脂代谢通路(Glycerophospholipid metabolism)、细胞周期代谢通路(Cell cycle)等。其中SMAD3、TLR2基因显著性富集在IBD代谢通路(见图9(a))。染色质易接近性减弱区域调控的表达下调基因显著性富集的代谢通路包括p53信号通路(p53 signaling pathway)、TRP通道炎症调控通路(Inflammatory mediator regulation of TRP channels)、半胱氨酸和蛋氨酸代谢通路(Cysteine and methionine metabolism)等。EI24基因显著性富集在p53信号通路(图9(b))。
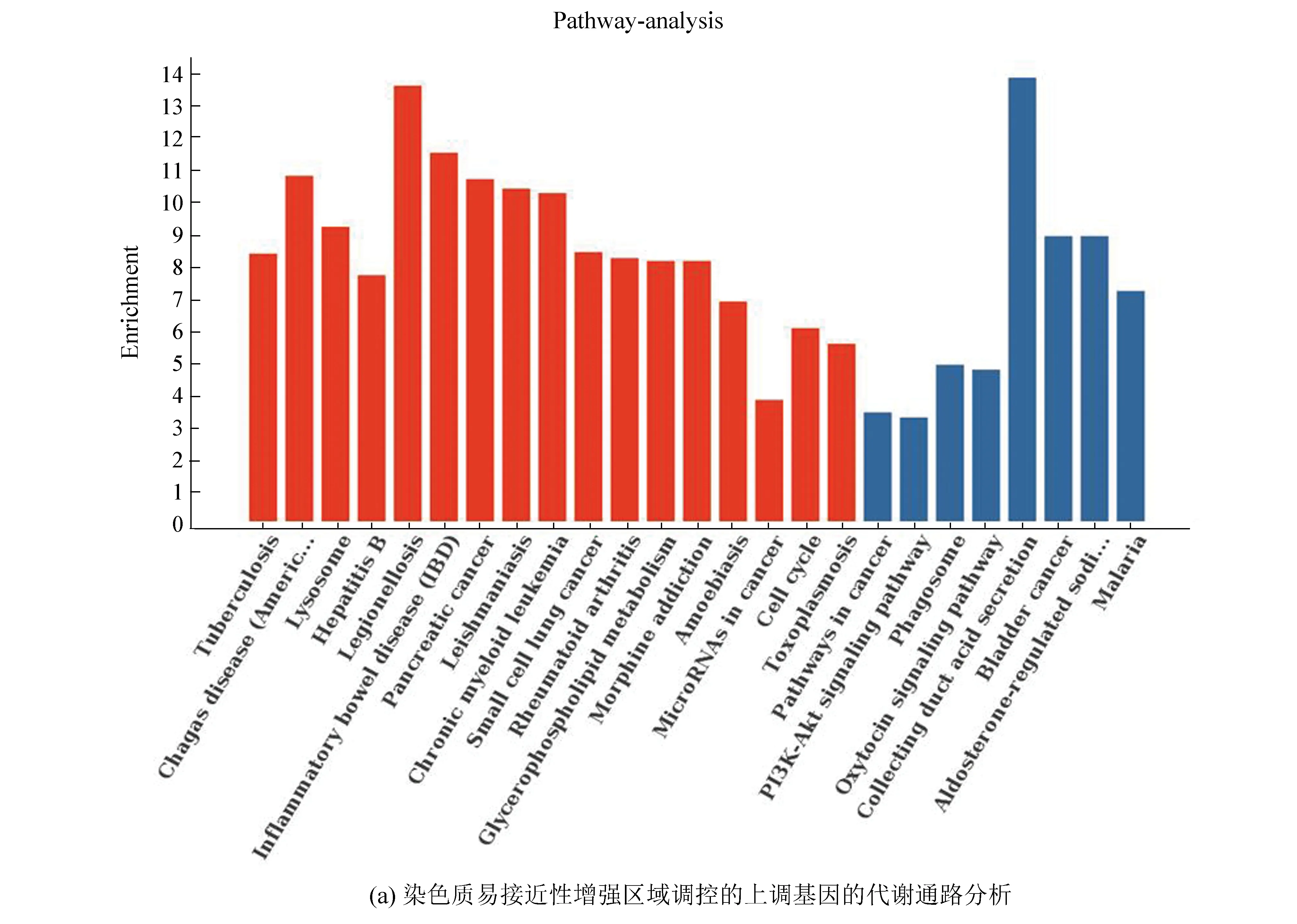
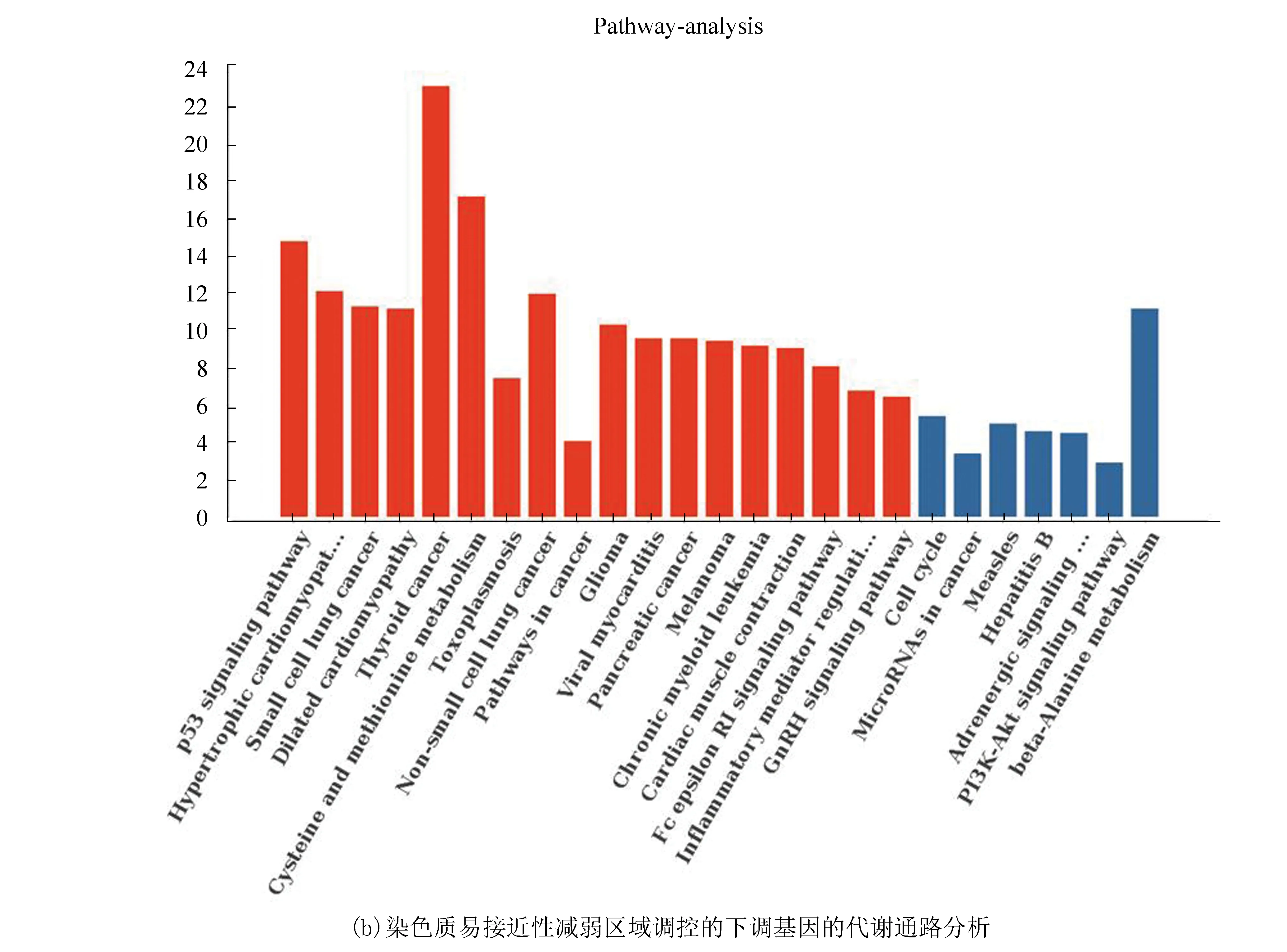
图9 代谢通路分析Fig.9 Metabolic pathway analysis
有研究表明,患有IBD的患者与健康人群相比患结直肠癌的风险更高[18-19],当肠道系统与其微生物群之间的关系(包括屏障功能、免疫信号和代谢物)受到干扰后引起的慢性炎症是其发病的主要潜在原因[20]。其次,在CRC中P53信号通路也是一种重要的信号通路,p53抑癌基因突变是导致CRC发生的最主要原因之一,同时也是结直肠癌侵袭和转移的原因之一,还有研究认为p53突变在腺瘤-癌转移过程中也发挥重要作用[21]。进一步研究IBD、P53信号通路在CRC发生发展中的作用将具有重要意义。
3 讨 论
一般来说,染色质有“关闭”“开放”两种状态,处于“关闭”状态的染色质,在异染色质蛋白以及修饰酶的作用下,被包装成致密、紧凑的结构,阻遏转录因子等蛋白的结合,此时染色质处于沉默失去生物功能的阶段;而处于“开放”状态的染色质,具有不太紧致的结构,可招募转录因子等蛋白的结合,进而调控基因的表达水平。ATAC-seq作为一种绘制全基因组染色质可及性图谱的方法[8],利用超活性Tn5转座酶检测染色质的可接近性,是本实验的重要研究手段之一[7]。通过对ATAC-seq和RNA-seq数据的分析,初步确定了FOXQ1基因敲低后发生差异表达的基因,丰富了FOXQ1转录因子的下游调控网络。
