高效液相色谱法测定复方依美辛胶囊中3种成分含量
2019-12-27郭社民
郭社民,王 静
(河南省濮阳市食品药品检验检测中心,河南 濮阳 457000)
复方依美辛胶囊是由马来酸依那普利、酒石酸美托洛尔、辛伐他汀、阿司匹林、氢氯噻嗪等组成的医院制剂,临床多用于治疗高脂血症、高血压、缺血性心脏病和脑卒中等症。原质量标准中仅对酒石酸美托洛尔进行了含量测定,为更有效地控制制剂质量,确保临床疗效,本研究中参考文献[1-10]建立了同时测定制剂中氢氯噻嗪、阿司匹林和酒石酸美托洛尔含量的高效液相色谱法。现报道如下。
1 仪器与试药
1.1 仪器
Agilent1260型高效液相色谱仪(美国Agilent公司);UVIKONxl型紫外分光光度计(法国Secoman公司);CPA225D型电子分析天平(德国 Sartorius公司);SK2200H型超声清洗器(上海科导超声仪器有限公司)。
1.2 试药
氢氯噻嗪对照品(批号为1000309-201404,含量99.7%),阿司匹林对照品(批号为100113-201706,含量99.8%),酒石酸美托洛尔对照品(批号为100084-201403),均购自中国食品药品检定研究院;复方依美辛胶囊(濮阳市第一人民医院制剂室,批号分别为20180226,20180421,20180619,规格为每粒含马来酸依那普利20 mg、酒石酸美托洛尔50 mg、辛伐他汀40 mg、阿司匹林75 mg、氢氯噻嗪12.5 mg);乙腈为色谱纯,其余试剂均为分析纯,水为纯化水。
2 方法与结果
2.1 色谱条件
色谱柱:XBridge C18柱(250 mm ×4.6 mm,5 μm);流动相:醋酸盐缓冲液(取醋酸铵3.9 g,加水810 mL溶解,加三乙胺 2.0 mL,冰醋酸10.0 mL,磷酸3.0 mL,摇匀)-乙腈(87 ∶13,V/V);流速:1.0 mL/min;检测波长:275 nm;柱温:35 ℃;进样量:20 μL。
2.2 溶液制备
混合对照品溶液:取氢氯噻嗪对照品15.25 mg、阿司匹林对照品90.19 mg、酒石酸美托洛尔对照品60.25 mg,精密称定,置同一20 mL容量瓶中,加甲醇-乙腈(1 ∶1,V/V)5 mL,超声 5 min,加流动相适量,超声(功率 100 W,频率 53 kHz,下同)处理 10 min,使其溶解,放冷,加流动相定容,摇匀,作为对照品贮备液(每1 mL含氢氯噻嗪0.760 2 mg、阿司匹林4.500 5 mg、酒石酸美托洛尔3.012 5 mg),量取5 mL,置100 mL容量瓶中,加流动相定容,摇匀,作为混合对照品溶液。
供试品溶液:取样品20粒,将内容物混匀,取适量(约相当于氢氯噻嗪7.6 mg),精密称定,置200 mL容量瓶中,加甲醇 -乙腈(1 ∶1,V/V)5 mL,超声处理5 min后加流动相适量,再超声处理10 min使其溶解,放冷,加流动相定容,摇匀,滤过,取续滤液,即得。
阴性对照品溶液:按复方依美辛胶囊处方比例及制备方法制备不含氢氯噻嗪、阿司匹林、酒石酸美托洛尔的阴性样品,并依法制备阴性对照品溶液。
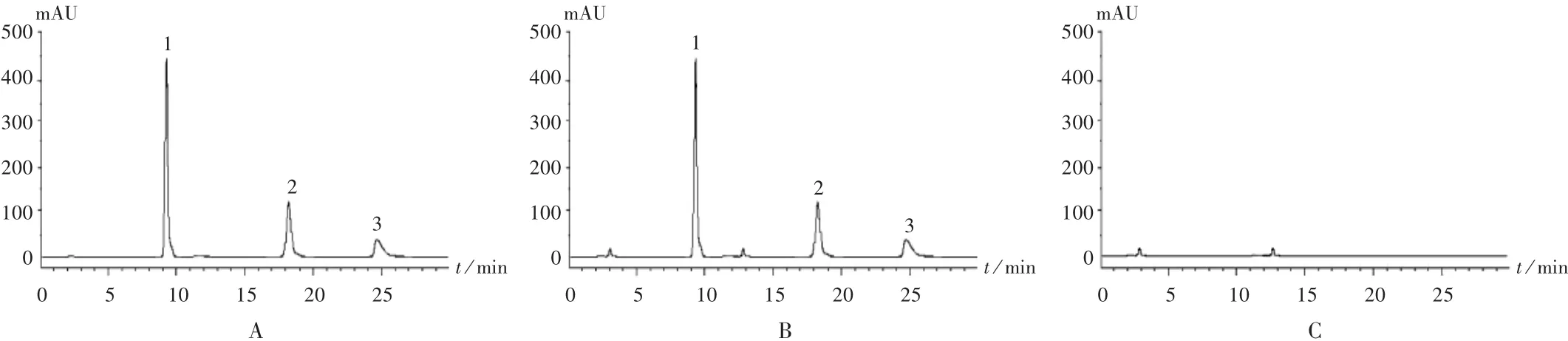
图1 高效液相色谱图
2.3 方法学考察
系统适用性试验:取上述混合对照品溶液、供试品溶液各适量,按拟订色谱条件进样测定。结果各待测成分峰形良好、分离度均大于1.5,理论板数以氢氯噻嗪、阿司匹林、酒石酸美托洛尔峰计均大于3000。色谱图见图1。
专属性试验:取对照品溶液、阴性对照品溶液各适量,按拟订色谱条件进样测定,记录色谱图,详见图1。结果,阴性对照品溶液在与混合对照品溶液保留时间相同处无干扰峰,表明方法专属性良好。
线性关系考察:分别精密量取混合对照品溶液0.4,0.6,0.8,1.0,1.5,2.0,2.5 mL,分别置 20 mL 容量瓶中,加流动相定容,摇匀,制成系列对照品溶液。取10 μL,按拟订色谱条件进样测定,记录峰面积。以待测成分质量浓度(X,mg/mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,得氢氯噻嗪、阿司匹林、酒石酸美托洛尔回归方程分别为Y1=1.346 1×105X1+42.13(r1=0.999 9,n=7),Y2=1.106 2×104X2+59.36(r2=0.999 9,n=7),Y3=9.187 5 ×103X3+86.52(r3=0.999 9,n=7)。结果表明,氢氯噻嗪、阿司匹林、酒石酸美托洛尔检测质量浓度分别在 0.015 2~0.095 0 mg/mL,0.090 0~0.562 6 mg/mL,0.060 2~0.376 6 mg/mL范围内与峰面积线性关系良好。
精密度试验:取混合对照品溶液适量,按拟订色谱条件连续进样6次,记录峰面积。结果氢氯噻嗪、阿司匹林、酒石酸美托洛尔峰面积的RSD分别为0.42%,0.48%,0.53%(n=6),表明仪器精密度良好。
稳定性试验:取同一供试品(批号为20180421)溶液,分别于室温下放置 0,2,4,6,8,10 h 时按拟订色谱条件进样测定,记录峰面积。结果氢氯噻嗪、阿司匹林、酒石酸美托洛尔峰面积的RSD分别为0.63%,0.71%,0.75%(n=6),表明供试品溶液在室温下放置10h内基本稳定。
重复性试验:取同一批(批号为20180421)样品,依法制备供试品溶液,共5份,再按拟订色谱条件进样测定,记录峰面积并计算含量。结果氢氯噻嗪、阿司匹林、酒石酸美托洛尔的平均含量分别为99.8%,101.3%,101.7%,RSD分别为 0.70%,0.64%,0.68%(n=5),表明方法重复性良好。
加样回收试验:取已知各成分含量的同一批(批号为20180421)样品内容物适量,共9份,分别加入低、中、高质量浓度的待测成分对照品溶液适量,依法制备供试品溶液,再按拟订色谱条件进样测定,记录峰面积,并计算回收率。结果见表1。
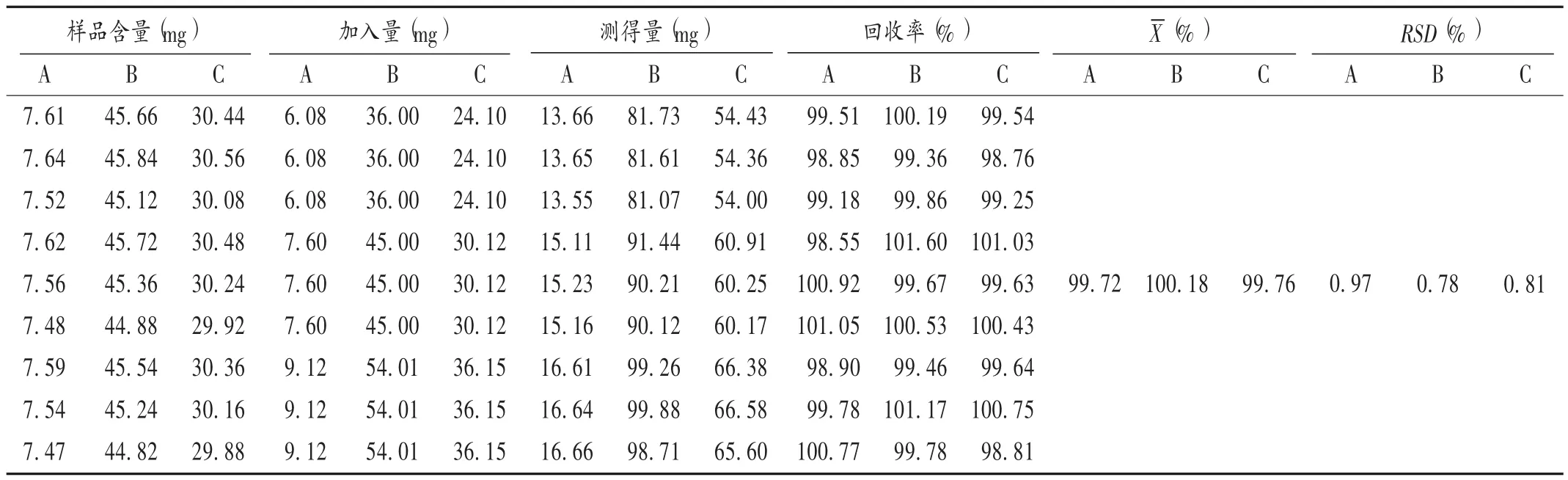
表1 加样回收试验结果(n=9)
2.4 样品含量测定
取样品适量,依法制备供试品溶液,再按拟订色谱条件进样测定,记录色谱峰面积并计算含量。结果见表2。批号20180226 20180421 20180619
3 讨论
溶剂选择:氢氯噻嗪在水中不溶解,阿司匹林微溶解,酒石酸美托洛尔极易溶解,三者的溶解性差异较大,氢氯噻嗪需要选择有机溶剂进行溶解。预试验结果表明,先加甲醇-乙腈(1∶1,V/V)适量溶解氢氯噻嗪后,再加流动相适量溶解其余2种成分,并进行超声处理,可使3种成分均得到充分溶解。
流动相选择:曾试用0.1 mol/L磷酸二氢钠-乙腈、醋酸盐缓冲液-甲醇、醋酸盐缓冲液-乙腈等系统及不同比例,试图用1种流动相同时分离和测定该制剂中各种主药成分,结果以文中溶液为流动相,能同时使氢氯噻嗪、阿司匹林、酒石酸美托洛尔3种成分很好地分离,且峰形较好、无干扰。
检测波长选择:在200~400 nm波长范围内,对氢氯噻嗪、阿司匹林、酒石酸美托洛尔对照品溶液分别进行全波长扫描,结果氢氯噻嗪、阿司匹林、酒石酸美托洛尔的最大吸收波长分别为273,276,275 nm,且酒石酸美托洛尔的响应值较小,故选用275 nm作为检测波长。
综上所述,本研究中所建方法简便、准确、重复性好,可用于复方依美辛胶囊中氢氯噻嗪、阿司匹林、酒石酸美托洛尔的含量测定。
