海洋天然产物Trichodermaxanthone分子光谱的理论研究
2019-12-20梁小蕊牛妍懿
梁小蕊,牛妍懿,刘 洁
(海军航空大学,山东烟台264001)
海洋生态环境较陆地生态环境具有高压、高盐、低氧、黑暗等特点。因此,海洋生物在新陈代谢、生存繁殖方式等许多方面具有与陆地生物不同的特性,能够代谢产生结构新颖和活性多样的天然产物,是研究发现新型药物的重要来源[1-6]。研究海洋天然产物非常重要的一个方面就是对分离纯化的单体化合物进行结构鉴定。随着现代仪器分析方法的不断发展和普遍应用,由紫外-可见光谱(UV-VIS)、红外光谱(IR)、核磁共振波谱(NMR)以及质谱(MS)为主的四种波谱分析方法相互配合,形成了一套完整的有机化合物结构鉴定方法。其中,红外光谱和核磁共振波谱具有很强的互补性,已成为测定各种有机、无机化合物分子结构的有力工具,但通常其谱峰较为复杂,不易分辨,而通过理论计算将光谱数据与实验数据相对比来进行分析,往往会得到事半功倍的效果[7-9]。
本文采用密度泛函理论(DFT),B3LYP 方法,在6-311G 基组水平上,对从泰国海扇Annella sp.来源的黄绿木霉(Trichoderma. aureoviride PSU-F95)次生代谢产物中分离得到的化合物trichodermaxanthone[10]进行了分子结构优化,在优化后的稳定结构基础上,计算了该化合物分子的红外光谱和核磁共振碳谱,将理论数据与实验数据对比分析,探讨谱峰的归属,为海洋天然产物的结构鉴定提供了理论依据。
1 计算方法
基于DFT的方法可以处理电子相关的问题,计算得到的分子结构和光谱数据等与实验数据吻合较好,并且比基于波函数的一些其他方法更为简单[11-16]。因此,本文采用DFT/B3LYP 方法,在6-311G 基组水平上,对trichodermaxanthone分子进行几何结构全优化,并通过计算振动频率来确认其结构的稳定性。以优化的稳定构型为基础,采用同样的方法,计算trichodermaxanthone的红外振动频率和核磁共振碳谱,全部计算利用Gaussian09软件包完成。
2 结果与讨论
2.1 分子的稳定构型
图1 给出了trichodermaxanthone 的平面分子结构,图2给出了利用DFT/B3LYP方法优化后的该分子的立体构型、原子编号及笛卡尔坐标。由图2 可看出trichodermaxanthone 分子中C1,C22,C13 所属的2 个苯环和1个六元环基本都在同一平面上,只有24号碳所在的取代基与苯环不在同一平面上。

图1 Trichodermaxanthone分子平面结构Fig.1 Planar structure of trichodermaxanthone
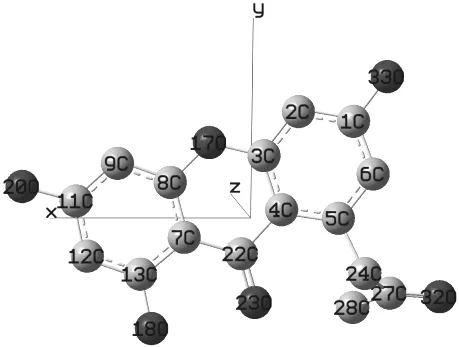
图2 优化后的trichodermaxanthone分子立体构型Fig.2 Optimized configuration of trichodermaxanthone
将优化构型的结构数据列入表1。
由表1 的二面角数据可知,化合物trichodermaxanthone 分 子 中 除 了∠C6—C5—C24—C27 =77.2° 、∠C5—C24—C27—C28 =68.1° 和∠C5—C24—C27—O32=111.8°外,其余二面角均在180°左右和0°左右,这说明除了C24所在的取代基与苯环不在同一平面外,其余各原子基本都在同一平面上。从表1 中的键角数据来看,所有键角都接近120°,这说明trichodermaxanthone分子中的各碳原子和氧原子都是sp2杂化方式成键。
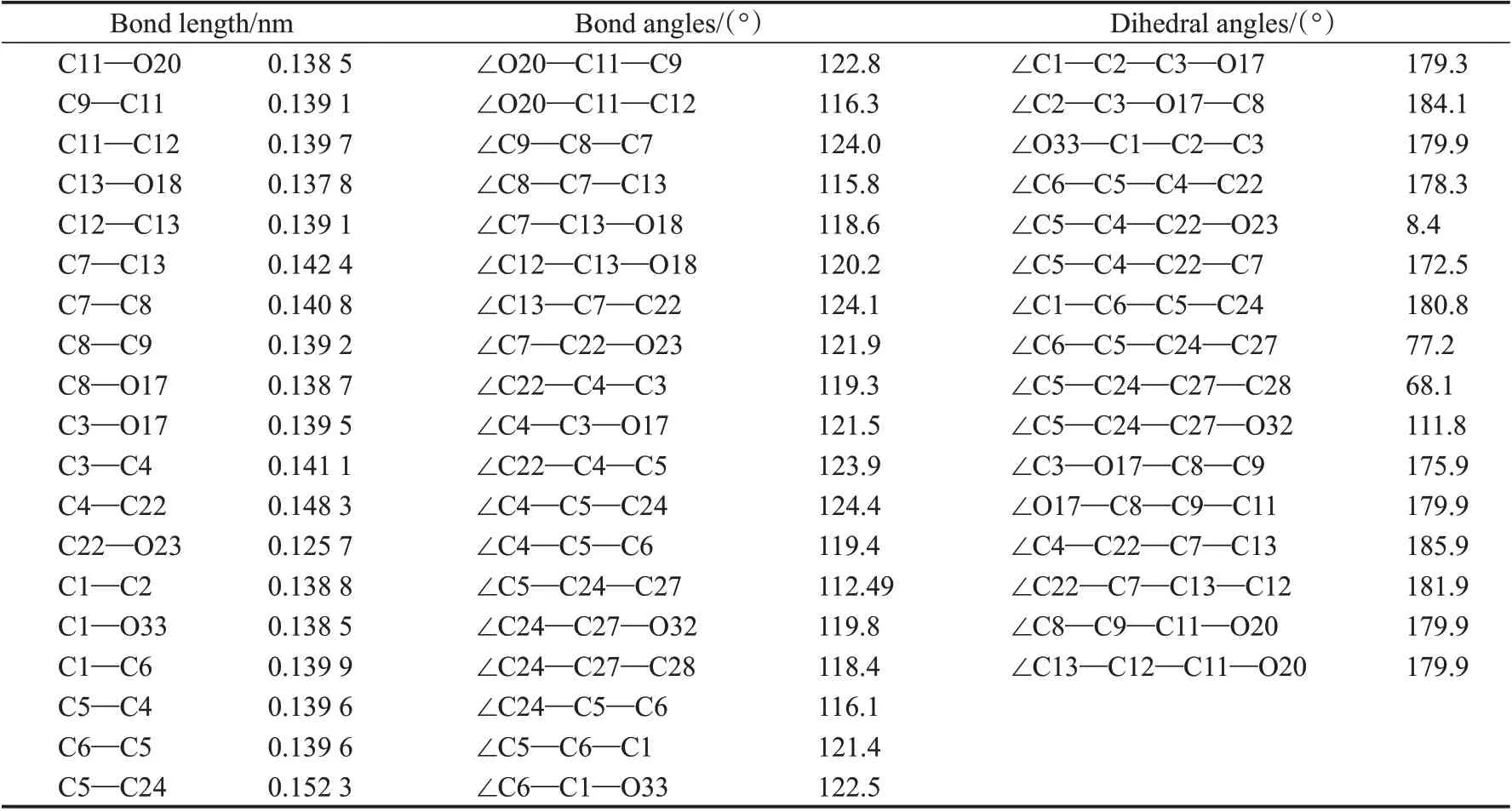
表1 优化后trichodermaxanthone分子的键长、键角、二面角数据Tab.1 Bond lengths and dihedral angles of trichodermaxanthone obtained by configuration optimization
表1 中的键长数据显示trichodermaxanthone 分子中的C11—O20 键长为0.138 5 nm、C13—O18 键长为0.137 8 nm 、C1—O33 键长为0.138 5 nm 均比单个苯酚上的碳氧单键0.143 0 nm 要短,分子中的苯环在靠近羟基一侧的碳碳键长较单个苯酚中的碳碳键长0.140 1 nm 略 短,如C9—C11、C11—C12、C1—C2、C6—C5 键 长 分 别 为 0.139 1 nm 、0.139 7 nm 、0.138 8nm、0.139 6 nm 等,而在靠近吡喃酮环的一侧键长则比单个苯酚中的碳碳键长要长,如C7—C13、C7—C8、C3—C4键长分别为0.142 4 nm、0.140 8nm、0.141 1 nm 等。从这些数据来看,取代基的存在使苯环形状有所扭曲,羟基的取代使苯环略有缩小,而靠近吡喃酮环的一侧苯环略有增大;从吡喃酮环来看,未取代的吡喃酮环其碳氧单键键长为0.143 6 nm、碳碳单键键长为0.154 1 nm、羰基处的碳碳单键键长为0.153 4 nm,而在trichodermaxanthone分子中这些键长变为:C8—O17 =0.138 7 nm 、C7—C8 =0.140 8 nm 、C4—C22=0.148 3 nm。可见,由于苯环的作用,使吡喃酮环缩小了。
2.2 前线分子轨道分析
用DFT,B3LYP 方法,在6-311G 基组水平上优化得到的 trichodermaxanthone 分子的能量为-1 068.22a.u.,即-29 067.76 ev,能量很低,优化构型达到最低能量状态。偶极矩为5.36 D,整个分子为极性分子。该分子最高占有轨道(HOMO)能量为-8.84 ev,最低空轨道(LUMO)能量为-5.71 ev,能隙为3.13 ev,主要是由于trichodermaxanthone 分子中存在离域大π 键,使得能隙较小,离域π 电子容易被激发,这可以从HOMO和LUMO电子云分布图中看出,如图3所示。

图3 Trichodermaxanthone分子的HOMO、LUMO图Fig.3 Highest occupied molecular orbital and lowest unoccupied molecular orbital of trichodermaxanthone molecule
2.3 分子红外光谱分析
利用DFT-B3LYP/6-311G方法计算出的trichodermaxanthone分子的红外振动光谱如图4所示。

图4 Trichodermaxanthone分子的红外谱图Fig.4 IR spectrum of trichodermaxanthone
按照红外吸收强度的不同,将分子的红外谱图分成3 部分,图4 中从上到下依次是380~1 000 cm-11 000~1 700 cm-1和3 000~3 700 cm-1范围的谱图。由于0~380 cm-1和1 700~3 000 cm-1这2个区域内几乎无红外吸收峰,因而不再讨论这2个区域。
380~1 000 cm-1区域内分子的振动模式主要是弯曲振动,其中面外弯曲振动产生的吸收峰更多,强度更强。397 cm-1、440 cm-1、812 cm-1、855 cm-1处均为面外弯曲振动模式,最强峰出现在397 cm-1处,是由21H 和34H 的面外弯曲振动中的垂直摇摆引起的,如图5所示;次强峰出现在440 cm-1处,是由19H的面外弯曲振动引起的;而这一区域内在381 cm-1和523 cm-1处出现的两个较弱峰主要是由18O、20O、33O 所在羟基的面内弯曲振动引起的,如图6所示。

图5 397 cm-1 处的振动模式Fig.5 Vibration mode of 397 cm-1

图6 523 cm-1 处的振动模式Fig.6 Vibration mode of 523 cm-1
1 000~1 700 cm-1范围内出现的红外吸收峰是整个分子中最多也是强度最强的部分,以面内弯曲振动模式居多。其中,在1 163 cm-1处出现的为最强吸收峰是由14H、10H、16H和3个羟基氢19H、21H、34H的面内弯曲振动引起的,如图7 所示;次强峰出现在1 640 cm-1处,与1 655 cm-1、1 671 cm-1处的2个红外吸收峰都是由分子中2个苯环的伸缩振动及其取代基的面内弯曲振动引起的,如图8 所示;1 037~1 088 cm-1处的几个较弱峰主要是由24C所在亚甲基和28C所在甲基的平面摇摆振动引起的。
3 000~3 700 cm-1范围内出现的谱峰是3个区域中最少,强度最弱的部分,这部分的振动模式均为伸缩振动。在3 673 cm-1、3 699 cm-1、3 703 cm-1处的3 个吸收峰分别由18O—19H、33O—34H 和20O—21H 的伸缩振动引起的;3 066 cm-1和3 094 cm-1处的吸收峰是由24C所在亚甲基的对称伸缩振动和28C所在甲基的不对称伸缩振动引起的。

图7 1 163 cm-1 处的振动模式Fig.7 Vibration mode of 1 163 cm-1

图8 1 640 cm-1 处的振动模式Fig.8 Vibration mode of 1 640 cm-1
2.4 分子核磁共振碳谱分析
核磁共振碳谱是研究处于强磁场中的碳核对射频辐射的吸收,从而获取有机化合物分子结构骨架信息。碳原子构成了有机化合物的骨架,因此,掌握有关碳原子的信息。在有机化合物结构鉴定中有重要意义[17-18]。本文采用DFT-B3LYP 方法计算得到的trichodermaxanth-one 分 子 的13C NMR 谱 见 图9。其中,158.0~161.5 ppm 范围内的碳信号较集中,无法分辨,因而图9 给出了这部分的放大图。13C NMR 谱显示trichodermaxanthone 分子有16 个碳信号,包括1 个甲基、1个亚甲基、6个次甲基和8个季碳。
表2 给出了trichodermaxanthone 分子碳谱的理论计算详细结果和文献[6]中的实验结果,理论数据显示:δc为22.7 ppm 和45.9 ppm 处的信号分别为28 号甲基和24号亚甲基的碳信号;分子中最大的碳信号出现在δc=217.3 ppm 处,是27号羰基碳所提供的信号;羟基取代在苯环上的3个季碳1C、11C、13C的碳信号分别出现在δc为159.5 ppm 、159.9 ppm 和160.9 ppm处。将理论数据和实验数据相比较发现除了27 号碳差值稍大外,其他原子碳信号的理论计算结果与实验结果吻合非常好,这说明采用的DFT-B3LYP/6-311G的计算方法是可行的,可以为化合物结构鉴定中的13C NMR 谱提供可靠的理论依据。理论数据和实验数据出现差别的主要原因是:实验碳谱是以氘代丙酮为溶剂进行测定的,而理论计算是在气相条件下进行的。

图9Fig.9 Calculated13C NMR Spectrum of Trichodermaxanthone

表2 Trichodermaxanthone分子核磁共振碳谱的理论计算结果和实验结果Tab.2 Calculated13C NMR data and experimental data of trichodermaxanthone ppm
3 结语
本文采用密度泛函DFT/B3LYP方法,从理论角度研究了一种从海洋木霉次生代谢产物中得到的化合物trichodermaxanthone。选取6-311G 基组,对trichodermaxanthone分子的结构进行了最低能量优化,分析了其优化构型的特点,发现该化合物各原子基本都在同一平面上,并且分子中的各碳原子和氧原子都是sp2杂化方式成键。从前线轨道分布图来看分子中存在离域大π 键。采用同样的方法和基组计算了trichodermaxanthone 分子的红外振动光谱,分析发现:1 000~1 700 cm-1范围内出现的红外吸收峰数目最多、强度最强,主要振动模式是面内弯曲振动;其次是380~1 000 cm-1区域,这一区域分子的振动模式以面外弯曲振动为主;3 000~3 700 cm-1范围内的振动模式均为伸缩振动,这一区域的谱峰是3个区域中最少,强度最弱的部分。对trichodermaxanthone分子碳谱的研究发现,理论计算与实验结果非常吻合。总的来说,本文的研究结果可为天然产物分子结构的鉴定提供光谱解析方面的理论依据,简化化合物结构鉴定的工作。
