混杂光固化3D打印树脂固化动力学性能
2019-12-17林广鸿尹敬峰黄伟滨蔡慕华向洪平刘晓暄
林广鸿,尹敬峰,黄 鸿,黄伟滨,蔡慕华,向洪平,刘晓暄
(广东工业大学 材料与能源学院,广州 510006)
环氧树脂是激光快速成型中常用的基础树脂,在3D打印耗材领域已有众多应用成果[1]。然而,环氧树脂成型速率慢,耐疲劳和耐冲击性能差,这使其应用受限[2]。聚氨酯丙烯酸酯中的丙烯酸酯基在UV辐照与自由基引发剂的作用下,能快速聚合形成三维交联网络,具有固化速率快、性能易调节等优点,在3D打印耗材领域也具有巨大应用潜力。但聚氨酯丙烯酸酯光固化过程中体积收缩大,导致固化制品易翘曲变形、成型精度低,阻碍了其发展与应用[3]。
以液态光敏树脂为基础的光固化3D打印技术,由于成型工艺简单、速率快、精度高,已受到广泛关注。自由基光固化体系速率快、性能易调节,但固化收缩严重,单独使用时成形精度低;阳离子固化体系黏度大速率慢,但体积收缩较小,能满足成形精度的要求[4]。自由基-阳离子混杂光固化体系能形成交联互穿网络(IPN),具有引发协同效应、性能互补等优势,能满足3D打印成型速率与精度的基本要求,且在3D打印领域具有重大应用前景[5]。郭长龙等[6]使用超支化聚酯丙烯酸酯预聚物,双酚A环氧树脂构建可自由基-阳离子混杂光固化的树脂体系以用于3D 打印制造柔性板材。Huang等[7]利用双酚A型环氧二丙烯酸酯,三丙二醇二丙烯酸酯,乙氧基化三甲氧基丙烷三丙烯酸酯和脂环族环氧树脂作为主体树脂形成自由基-阳离子混杂光固化体系,可满足印刷及3D 打印要求。但对于3D打印用自由基-阳离子混杂光固化树脂的固化动力学及其性能调控还鲜见报道。
为此,本工作拟以环氧树脂(EPON828)和聚氨酯丙烯酸酯(RJ429)作为主体树脂来制备自由基-阳离子混杂光固化树脂;利用实时红外(real-time FT-IR)监控光固化过程中丙烯酸酯基的双键和环氧基的转化率,优化混杂光固化体系中自由基和阳离子光引发剂的种类,质量配比和加入量;并进一步通过比较在自由基/阳离子光引发剂的最佳质量配比和加入量下光敏树脂的黏度及其光固化器件的力学性能,体积收缩和翘曲度等性能,确定环氧树脂和聚氨酯丙烯酸酯的最佳比例,以此获得成型速率快、成型精度高的可用于3D打印的光固化树脂。
1 实验材料与方法
1.1 试剂与仪器
试剂:双酚A型环氧树脂828(EPON828)和聚氨酯丙烯酸酯(RJ429)均为工业级。自由基光引发剂TPO, Irgacure 369, Irgacure 651, Irgacure 184, Doracur 1173;阳离子光引发剂Gencure 842, Gencure 831, Gencure 202,均为工业级。
仪器:傅里叶变换红外光谱仪(iS50, FT-IR);万能材料试验机(ANS);悬臂梁冲击试验机(XJU);紫外光照射装置(MUA-165),光源波长范围320~400nm,主波长365nm。
1.2 测试与表征
通过实时红外(real-time FT-IR),检测聚氨酯丙烯酸酯中丙烯酸酯双键峰(1640cm-1)和环氧树脂中的环氧峰(930cm-1),按公式(1)和公式(2)分别计算双键和环氧基的转化率[8]。根据转化率增长快慢比较基团的聚合速率大小。
(1)
(2)
式中:W为基团转化率;(A1640/A2940)0,(A930/A2940)0为双键或环氧基光固化反应前的峰面积;(A1640/A2940)t,(A930/A2940)t为双键或环氧基光固化反应后的峰面积。
固化深度测定:将混杂光固化树脂置于10mm×10mm×3mm的凹槽中,光照5s后将固体部分取出,洗去残余预聚体,测量厚度。固化表干时间测定:参照GB/T 1728-1979测试标准进行测定。拉伸强度测试:参照ASTM D638-2014进行拉伸测试,拉伸速率为10mm/min。冲击强度测试:参照GB/T 1843-2008测试标准进行测试。硬度测试:参照GB/T 2411-1980测试标准进行测试。凝胶含量测试:将混杂光固化树脂经UV固化后,称量固化树脂(m1/g),浸泡甲苯中48h干燥后称量质量(m2/g),按公式(3)计算凝胶率G。
(3)
体积收缩率测试:参照国际标准ISO 3521-1997中的密度法进行测试。翘曲度测试:翘曲度是对材料翘曲程度的判定,按公式(4)计算翘曲度[9],测试示意图如图1所示。
γ=h/L
(4)
式中:γ为塑件特定部分翘曲度;h为翘曲量;L为评价目标在特定方向的投影长度。

图1 翘曲度测试示意图Fig.1 Schematic diagram of warpage test
2 结果与分析
2.1 混杂光固化树脂的聚合原理
双酚A型环氧树脂828(EPON828)和聚氨酯丙烯酸酯(RJ429)在自由基/阳离子光引发剂的作用下,两种聚合物经UV辐照后形成交联互穿网络(IPN)结构,表现出优良的综合性能,过程图如图2所示。

图2 混杂光固化树脂的聚合过程Fig.2 Photopolymerization of hybrid UV-curing resin
2.2 引发剂的选择
2.2.1 阳离子光引发剂的选择
为考察不同阳离子光引发剂对EPON 828的引发固化效果,选取3种常用的阳离子光引发剂(Gencure 842,Gencure 831和Gencure 202),所用引发剂吸收峰和最大吸收波长如表1所示,并分别以3%(质量分数,下同)的比例加入环氧树脂828中;然后在40mW·cm-2的辐照强度下UV固化,用实时红外对环氧树脂中环氧基的转化率随辐照时间变化进行原位监测。图3为不同阳离子光引发剂引发环氧树脂828光固化反应的转化率测试结果。由图3可知,Gencure 831, Gencure 842和Gencure 202光引发环氧基聚合的最大转化率分别为59.1%, 64.5%和67.0%;其中Gencure 202光引发环氧基聚合的聚合速率最快,Gencure 842引发聚合速率相对较慢。表2为测试不同阳离子光引发剂下环氧树脂828固化性能的结果。由表2可知,Gencure 842引发环氧树脂固化的固化深度最大(3.21mm),表干时间最短(10s),拉伸强度(3.89MPa)和冲击强度(2.65kJ·m-2)最高,这是由于Gencure 842引发聚合速率相对较慢,有助于深层固化[10]。虽然Gencure 842引发聚合速率慢于Gencure 831和Gencure 202,但最大转化率与Gencure 202接近,高于Gencure 831。综合考虑聚合过程与固化制品性能,选择Gencure 842作为混杂光固化体系中环氧树脂的光引发剂比较合适。

表1 阳离子引发剂的吸收峰范围和最大吸收波长Table 1 Absorption peak range and maximum absorption λmax of cationic photoinitiators

图3 不同阳离子光引发剂引发环氧树脂828光固化反应的转化率Fig.3 Conversion rate of UV-curing reaction in epoxy resin 828 initiated by different cationic photoinitiators

表2 不同阳离子光引发剂下环氧树脂828固化后的性能Table 2 Performances of UV-cured epoxy resin 828 with different cationic photoinitiators
选择合适阳离子光引发剂后,需进一步考察引发剂加入量对环氧树脂的引发固化效果。将阳离子光引发剂Gencure 842分别以1%~6%(质量分数,下同)加入量加入环氧树脂828中,在40mW·cm-2辐照强度下UV固化,用实时红外对环氧树脂中环氧基的转化率随辐照时间的变化进行原位监测。图4为不同842加入量引发环氧树脂828光固化反应的转化率测试结果。由图4可知,随着Gencure 842的加入量增大,引发环氧基聚合转化率和反应速率呈上升趋势,加入量增大一定程度后,转化率和反应速率上升程度不大甚至出现下降趋势。这种现象主要是由笼蔽效应引起,引发剂加入量过多,界面引发剂可以引发单体或和内部引发剂发生副反应,内部引发剂不能接触单体而只能发生副反应导致引发效率降低[11]。当Gencure 842加入量为5%时,环氧基转化率最高,可达90.7%。综合考虑转化率与反应速率,可确定阳离子光引发剂Gencure 842引发环氧树脂828光固化的最佳加入量为5%。

图4 不同Gencure 842加入量引发环氧树脂828光固化反应的转化率Fig.4 Conversion rate of UV-curing reaction in epoxy resin 828 initiated by different dosage of Gencure 842
2.2.2 自由基光引发剂的选择
为考察自由基光引发剂对聚氨酯丙烯酸酯树脂RJ429的引发固化效果,选取常用的5种自由基光引发剂(Irgacure 651, TPO, Doracur 1173, Irgacure 184和369),所用引发剂吸收峰和最大吸收波长如表3所示。并分别以3%加入量加入聚氨酯丙烯酸酯树脂RJ429中;在40mW·cm-2辐照强度下UV固化,用实时红外对聚氨酯丙烯酸酯树脂中双键的转化率随辐照时间的变化进行原位监测。图5为不同自由基光引发剂引发聚氨酯丙烯酸树脂RJ429光固化反应的转化率测试结果。由图5可知,Doracur 1173引发双键聚合反应的转化率最高,达到70.2%,369次之,而Irgacure 184和TPO接近,约为54%,Irgacure 651最低。此外,369引发双键反应速率最快,TPO次之,Irgacure 184和1173引发速率接近,651最慢。

表3 自由基光引发剂的吸收峰范围和最大吸收波长Table 3 Absorption peak range and maximum absorption λmax of free radical photoinitiators

图5 不同自由基光引发剂引发聚氨酯丙烯酸树脂RJ429光固化反应的转化率Fig.5 Conversion rate of UV-curing reaction in polyurethane acrylate RJ429 initiated by different free radical photoinitiators
比较各自由基光引发剂的引发效果要兼顾与阳离子光引发剂的复配协同效应,因此,选择合适的自由基光引发剂还需考察与阳离子光引发剂复配后的引发效果。阳离子光引发剂和自由基光引发剂复配比例通常控制在2∶1[12]。将阳离子光引发剂Gencure 842和5种自由基光引发剂(Irgacure 651, TPO, Doracur 1173, Irgacure 184和369)以质量配比为2∶1,加入量为2%组成复配引发剂加入环氧树脂828中,在辐照强度为40mW·cm-2下UV固化,用实时红外对环氧树脂中环氧基的转化率随辐照时间的变化进行原位监测。图6为Gencure 842与不同自由基光引发剂复配引发环氧树脂828光固化反应的转化率测试结果。由图6可知,与单独用Gencure 842引发环氧基聚合相比,369和TPO的加入抑制Gencure 842对环氧基聚合的转化率;Irgacure 651的加入提高了Gencure 842对环氧基聚合的转化率,但降低了其聚合速率;Irgacure 184和Doracur 1173的加入不仅促进了Gencure 842对环氧基聚合的转化率,还提高了其聚合速率。Irgacure 184促进Gencure 842引发的效果最佳,环氧基转化率达56%,而Doracur 1173促进Gencure 842引发的效果次之,环氧基转化率达52.7%。

图6 Gencure 842与不同自由基光引发剂复配引发环氧树脂828光固化反应的转化率Fig.6 Conversion rate of UV-curing reaction in epoxy 828 initiated by Gencure 842 compounded with different free radical photoinitiators
复配引发剂要兼顾复配光敏树脂中环氧基和双键在固化时的转化率和反应速率[13]。综上所述,Irgacure 184促进Gencure 842引发的效果略高于Doracur 1173,但Irgacure 184对双键的引发效果不佳,因此选择Doracur 1173作为复配引发剂的自由基光引发剂。
选择合适的自由基光引发剂后,还需单独考察引发剂加入量对聚氨酯丙烯酸酯树脂的引发固化效果。将自由基光引发剂Doracur 1173分别以1%~6%加入量加入聚氨酯丙烯酸酯树脂RJ429中,在辐照强度40mW·cm-2下进行UV固化,用实时红外对聚氨酯丙烯酸酯树脂中双键的转化率随辐照时间的变化进行原位监测。图7为不同Doracur 1173加入量引发丙烯酸树脂429光固化反应的转化率测试结果。由图7可知,随着Doracur 1173加入量增大,引发双键聚合的转化率和反应速率呈上升的趋势,而加入量增大到一定程度后,转化率和反应速率上升程度不大甚至下降,这种现象也是笼蔽效应引起的。当Doracur 1173加入量为5%时,引发双键聚合转化率最高,达到89.9%,反应速率最快。因此,选用自由基光引发剂Doracur 1173引发双键聚合的最佳加入量为5%。

图7 不同Doracur 1173加入量引发聚氨酯丙烯酸树脂RJ429光固化反应的转化率Fig.7 Conversion rate of UV-curing reaction in polyurethane acrylate RJ429 initiated by different dosage of Doracur 1173
2.3 复配光引发剂质量配比及加入量的确定
根据上述结果,将复配光引发剂中阳离子/自由基质量配比(C/R)分别设置为0.50,0.75,1.00,1.25和1.50,预配混杂光固化树脂中环氧树脂与聚氨酯丙烯酸酯树脂的比例为1,以复配光引发剂加入量为5%加入混杂光固化树脂中,在辐照强度40mW·cm-2下UV固化,用实时红外对光敏树脂中双键和环氧基的转化率随辐照时间的变化进行原位监测。图8为不同复配光引发剂质量配比引发双键和环氧基团反应的转化率测试结果。由图8可知,双键和环氧基转化率随着C/R值增大先升高后降低,反应速率随着C/R值增大先增快后减慢。当质量配比为0.75时,双键转化率最高,可达95.4%,环氧基转化率仅次于质量配比为1时的环氧基转化率,达74.7%。当质量配比为1时,环氧基转化率最高可达81.0%,双键转化率达84.9%。综合考虑双键与环氧基的转化率和反应速率以及引发剂成本,选择C/R值为0.75。

图8 不同复配光引发剂质量配比引发双键反应的转化率(a)和环氧基团反应的转化率(b)Fig.8 Conversion rate of reaction of double bond (a) and epoxy group (b) initiated by different ratio of hybrid photoinitiators
将复配光引发剂以质量配比为0.75,加入量为3%~7%加入预配混杂光固化树脂中,在40mW·cm-2辐照强度下UV固化,用实时红外对光敏树脂中双键和环氧基的转化率随辐照时间的变化进行原位监测。图9为不同复配光引发剂加入量引发双键和环氧基团反应的转化率测试结果。由图9可知,随着复配引发剂加入量增加,双键和环氧基的转化率和反应速率先上升后下降,这也是笼蔽效应引起的。当加入量为6%时的双键和环氧基的转化率分别为93.3%和81.4%,具有相当高的反应速率,因此,复配引发剂的加入量选用6%。

图9 不同复配光引发剂加入量引发双键反应的转化率(a)和环氧基团的反应转化率(b)Fig.9 Conversion rate of reaction of double bond (a) and epoxy group (b) initiated by different dosage of hybrid photoinitiators
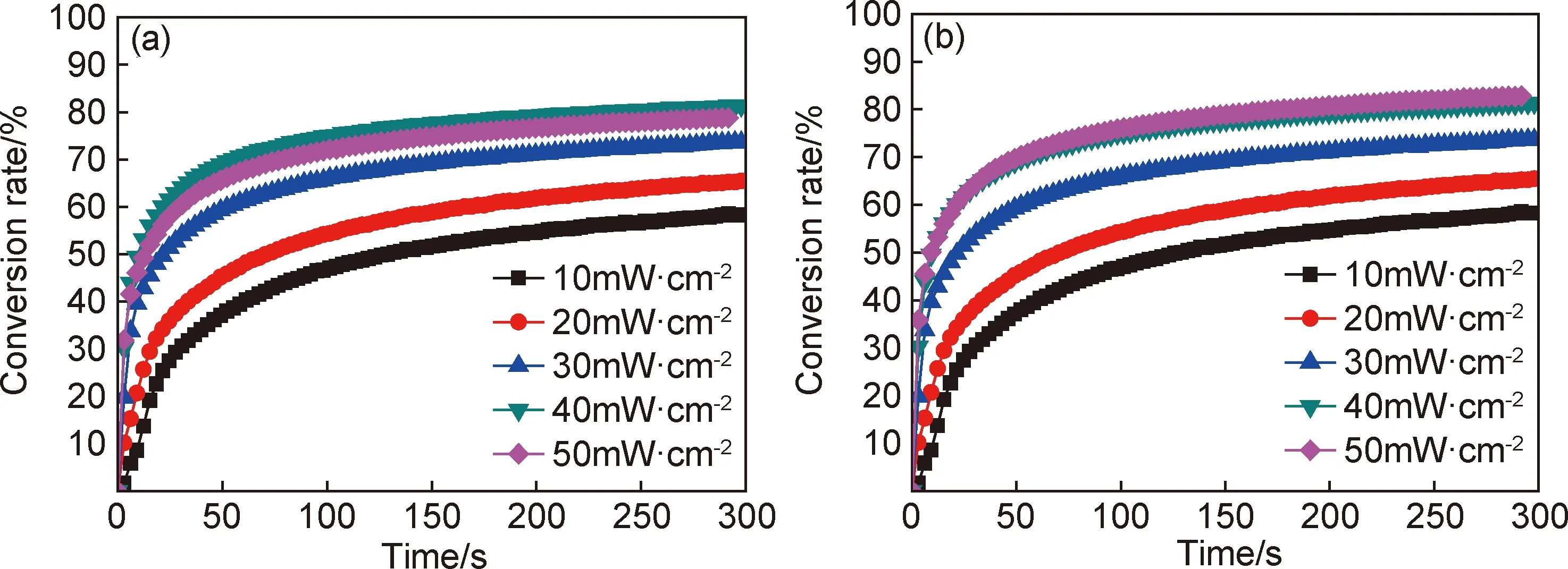
图10 不同辐照强度下双键反应的转化率(a)和环氧基团反应的转化率(b)Fig.10 Conversion rate of reaction of double bond (a) and epoxy group (b) under different radiation intensity
2.4 辐照强度的选择
将复配光引发剂以质量配比0.75,加入量6%加入预配混杂光固化树脂中,分别在10~50mW·cm-2辐照强度下UV固化,研究辐照强度对光固化过程的影响。用实时红外对光固化树脂中双键和环氧基的转化率随辐照时间的变化进行原位监测。图10为不同辐照强度下双键和环氧基团反应转化率测试结果。由图10可知,随着辐照光强的升高,双键和环氧的反应转化率和反应速率上升,辐照光强的升高,促使单位面积引发剂吸收更多光能,加快引发剂分解效率,产生更多自由基和阳离子,有利于反应转化率和反应速率的提高[14]。综合转化率,反应速率和节约能耗三方面考虑,混杂光固化树脂在40mW·cm-2和50mW·cm-2辐照强度下,反应基团的转化率和反应速率相差较小,因此辐照强度选择40mW·cm-2。
2.5 混杂光固化树脂复质量配比例的选择
在光固化过程中,环氧树脂与聚氨酯丙烯酸酯树脂质量配比(E/A)影响混杂光固化树脂的性能。将E/A分别设置为0, 0.50, 0.75, 1.00, 1.50和2.00,测其黏度,在辐照强度40mW·cm-2下,用引发剂质量配比0.75,加入量6%的复配光引发剂对不同混杂光固化树脂UV固化,并对固化树脂的凝胶含量、硬度、拉伸强度、冲击强度、体积收缩率和翘曲度进行测试,综合各方面性能,选择合适的复配光固化树脂质量配比。
2.5.1 混杂光固化树脂的黏度
图11为光固化树脂黏度测试结果。由图11可知,环氧树脂828与丙烯酸树脂429以及两种树脂复配的光固化树脂都具有剪切变稀的特性。此外,E/A值增加,复配体系光敏树脂的黏度随之上升。环氧树脂黏度较大影响树脂流动性,在加入聚氨酯丙烯酸酯后,复配体系黏度有所降低。

图11 环氧树脂828,丙烯酸树脂429与混杂光固化树脂的黏度Fig.11 Viscosity of epoxy resin 828, acrylic resin 429 and hybrid UV-curing resin
2.5.2 混杂光固化树脂的力学性能
图12为光固化树脂凝胶含量与硬度测试结果。由图12可知,E/A值为1.00时,凝胶含量最高,达到86.8%,其硬度仅次于质量配比为0.50,达到78 A。图13为光固化树脂力学性能测试结果。由图13可知,随E/A值的增加,混杂光固化树脂固化后的拉伸强度降低,冲击强度增强,这是因为环氧树脂具有良好的硬度,聚氨酯丙烯酸酯树脂具有良好的柔韧性[15];当E/A值增加后,环氧树脂的硬度提高了固化后树脂的冲击强度,但同时一定程度上掩盖了聚氨酯丙烯酸酯树脂的柔韧性,降低了固化后树脂的拉伸强度。质量配比为0.50时,固化样条的拉伸强度最高,达到8.38MPa;质量配比为1.50时,固化样条的冲击强度最高,达到8.81kJ·m-2;质量配比为1.00时,固化样条的拉伸强度和冲击强度分别6.60MPa和8.28kJ·m-2。

图12 混杂光固化树脂的凝胶质量分数与硬度Fig.12 Gel mass fraction and hardness of the hybrid UV-cured resin
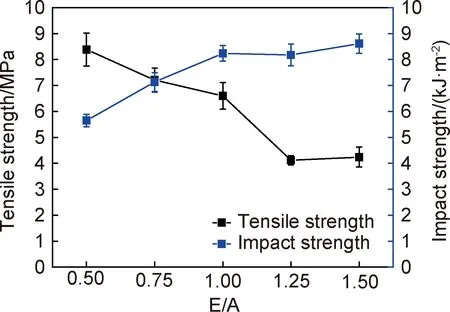
图13 混杂光固化树脂的拉伸强度与冲击强度Fig.13 Tensile strength and impact strength of the hybrid UV-cured resin
2.5.3 混杂光固化树脂的成型精度
树脂的成型精度主要由固化后的体积收缩和翘曲情况判断,体积收缩率和翘曲度越低,成型精度越高[16]。将混杂光固化树脂固化成80mm×10mm×4mm的样条,测量树脂固化前后的密度变化,得出体积收缩情况,并测量翘曲度。图14为固化后的透明样条。如图14所示,样条表面光泽,与纯聚氨酯丙烯酸酯(E/A=0)相比,样条翘曲情况明显降低。表4是混杂光固化树脂固化后的体积收缩和翘曲情况。由表4可知,随着E/A值的增加,树脂的体积收缩率下降,这是因为环氧基聚合产生的收缩应力较小,弥补了双键的聚合收缩,使得固化后体积收缩率下降。环氧基含量越高,体积收缩率越低,成型精度越高,这是环氧-丙烯酸酯复配体系独有的“体积互补效应”所造成的[17]。当E/A值升到1.00以后,体积收缩变化不大,这是因为当环氧树脂比例提高到成为体系中大部分时,丙烯酸树脂固化效果对整体树脂的影响逐渐变小,而环氧基在丙烯酸酯存在情况下,固化性能在很大程度上取决于丙烯酸酯树脂中双键的转化程度[18]。光敏树脂的固化翘曲度主要与形成交联网络时压力释放有关,若交联固化过程中交联网络处于高度无序状态,分子内部压力释放受阻,树脂将发生翘曲变形[19]。随着E/A值的增加,树脂的翘曲度先下降后上升,这是因为环氧树脂能与丙烯酸酯形成交联互穿网络(IPN),使交联网络整合有序,翘曲度明显变低;当E/A值升到1.00以后,翘曲度略微上升,这是因为环氧树脂提高到成为体系中大部分时,分子内部交联互穿网络(IPN)受到环氧树脂交联网络的影响,无序度增大,内部释放压力受阻,翘曲度上升[20]。

图14 混杂光固化树脂固化样条Fig.14 Spline of hybrid UV-cured resin

E/AVolume shrinkage/%Warpage/%0-7.27811.270.50-5.0725.780.75-4.5595.061.00-3.9863.621.50-3.9274.122.00-3.7893.97
综上所述,在质量配比为1.00时,混杂光固化树脂固化后的硬度,冲击强度和拉伸强度均较为优异,黏度为50.5 Pa·s,且体积收缩率和翘曲度较低,因此混杂光固化树脂的复配比例选用1.00。
3 结论
(1)以环氧树脂828和聚氨酯丙烯酸酯树脂RJ429为基础树脂制备了一种自由基-阳离子混杂光固化3D打印树脂。
(2)环氧树脂828选用阳离子引发剂Gencure 842最为适宜,最佳加入量为5%;自由基引发剂Doracur 1173和Irgacure 184促进Gencure 842的引发效果,Irgacure 651不影响Gencure 842的引发效果,而369和TPO会抑制Gencure 842的引发效果。聚氨酯丙烯酸酯树脂RJ429选用Doracur 1173最为适宜,最佳浓度为5%。
(3)阳离子引发剂Gencure 842和自由基引发剂Doracur 1173复配引发剂的质量配比选择为0.75,加入量为6%。环氧树脂828和聚氨酯丙烯酸酯树脂RJ429的复质量配比例为1.00。
(4)混杂光固化树脂的黏度为50.5Pa·s,拉伸强度和冲击强度分别6.60MPa和8.28kJ·m-2,体积收缩率和翘曲度分别为-3.986%和3.62%,满足3D打印要求。
