Eight key long non-coding RNAs predict hepatitis virus positive hepatocellular carcinoma as prognostic targets
2019-12-14ZiLinHuangWangLiQiFengChenPeiHongWuLuJunShen
Zi-Lin Huang,Wang Li,Qi-Feng Chen,Pei-Hong Wu,Lu-Jun Shen
Zi-Lin Huang,Wang Li,Qi-Feng Chen,Pei-Hong Wu,Lu-Jun Shen,Department of Medical Imaging and Interventional Radiology,Sun Yat-sen University Cancer Center,Guangzhou 510060,Guangdong Province,China
Zi-Lin Huang,Wang Li,Qi-Feng Chen,Pei-Hong Wu,Lu-Jun Shen,State Key Laboratory of Oncology in South China,Collaborative Innovation Center for Cancer Medicine,Guangzhou 510060,Guangdong Province,China
Abstract
Key words: Long non-coding RNAs; Hepatitis virus; Hepatocellular carcinoma;Prognostic signature; Least absolute shrinkage and selection operator
INTRODUCTION
Hepatocellular carcinoma (HCC),one of the most frequent liver cancer types,is a severe worldwide health problem[1].Chronic hepatitis B virus (HBV) infection and/or hepatitis C virus (HCV) infection have been identified as the leading causes of HCC[2].Specifically,the prevalence of HCV infection is predominant in European countries,which accounts for about 60% of all HCC cases in Italy and Spain; by contrast,HBV infection is more commonly seen in eastern Asian countries such as China,with an incidence rate of over 2/3[3].Specifically,hepatitis viruses have been epidemiologically associated with the development of HCC[4,5].
It has been extensively determined that hepatitis viruses are associated with HCC etiologically; nonetheless,it is still not completely clear about the molecular mechanism of hepatitis viruses in inducing liver cancer.An increasing number of mutation genes,like TP53,CTNNB1,and mTOR[6],are found to participate in HCC genesis as well as progression.Nonetheless,HCC is a highly heterogeneous disease,which can determine the altered biological progression of HCC and add to the difficulty in predicting prognosis.As a result,it is urgently needed to search for the efficient prognostic biomarkers for HCC.
Long non-coding RNAs (LncRNAs) have been identified to be the superior prognostic biomarkers over other cancer hallmarks,which can be ascribed to their unique advantages,as shown below.First,lncRNA expression is greatly distinct among different tissues,in various diseases,as well as at different disease progression stages; as a result,they can better represent the disease characteristics[7].Second,lncRNAs have been found to regulate the expression of genes at the transcriptional,post-transcriptional,and epigenetic levels[8,9]; on this account,their levels as well as functions show a close association with cancer progression.As a result,increasing studies have been carried out to illustrate the clinical value of lncRNAs among cancers,such as HCC[10].
In this study,hepatitis virus positive HCC (VHCC) patients were appropriately selected to explore the difference in the lncRNA expression profiles,so as to identify the candidate lncRNA biomarkers based on The Cancer Genome Atlas (TCGA)database.Moreover,the key lncRNAs were determined using the least absolute shrinkage and selection operator (LASSO) algorithm; thereafter,the risk score system for VHCC was established.This study aimed to establish a potent lncRNA signature based on the expression profiles through thorough genomic data analysis,thus improving the accuracy in predicting the prognosis of VHCC.
MATERIALS AND METHODS
Patient datasets as well as processing
The data were downloaded from TCGA database.Gene expression,together with the clinical information of HCC patients (https://tcga-data.nci.nih.gov/),was downloaded using the Data Transfer Tool (provided by GDC Apps).Among them,151 patients were virus-related.Patients with unknown lncRNA expression were eliminated from this study (n= 2); as a result,a total of 149 HCC cases were finally enrolled for analysis.The flowchart of analysis is shown in Figure1.A total of 149 virus-induced HCC cases were enrolled for comprehensive integrated analysis.The data were publicly available,so approval was exempted from the Ethics Committee.Data were processed in accordance with the data access policies as well as the NIH TCGA human subject protection policies (http://cancergenome.nih.gov/publications/publicationguidelines).
LncRNA expression in HCC patients was derived from Illumina HiSeq RNASeq platform,which was then standardized based on TCGA.Then,the R software“edgeR” package was employed to detect the differentially expressed RNAs,so as to identify the differentially expressed lncRNAs (DElncRNAs),and the thresholds were set at log2fold change > 2.0 as well as adjustedPvalue < 0.05.
Construction of lncRNA signature
The relationship of each lncRNA expression with the overall survival (OS) of patients was calculated using the univariate Cox model.Typically,lncRNAs would be deemed as statistically significant atP< 0.05 upon univariate Cox analysis.Subsequently,the chosen lncRNAs were screened and verified through LASSO regression with the R software “glmnet” package.Finally,the lncRNA-based prognostic risk score was constructed by linearly combining the products of expression level with the regression model (β) according to the formula below:Risk_score = βlncRNA1 ×lncRNA1 expression + βlncRNA2 × lncRNA2 expression + · ···· + βlncRNAn ×lncRNAn expression.
Confirmation of lncRNA signature
Patients together with their survival information were distributed according to the risk_score.Furthermore,patients were classified into a high-risk group and low-risk group according to the median risk_score.Then,the Kaplan-Meier survival curves were plotted,which could predict the high or low risk of patients.Thereafter,the univariate Cox proportional hazards regression analysis was performed.Then,the sensitivity and specificity regrading survival prediction were compared by the risk_score,and the receiver operating characteristic (ROC) curves with time dependence was then employed to evaluate the accuracy in predicting the 5-year prognosis.Moreover,multivariate Cox regression was also performed to examine whether lncRNA risk_score was predicted independently from other clinical factors.The R-package “party” “ctree” function was used to construct a conditional inference tree for better illustration.Besides,a nomogram was also established with the R project “rms” package.In addition,the predictive performance of the nomogram was verified for discrimination and calibration.Subsequently,subgroup analysis based on virus type was carried out.All the two-sidedP< 0.05 would be deemed to be statistically significant.The R software (version 3.4.1,R Foundation) was employed for all analyses.
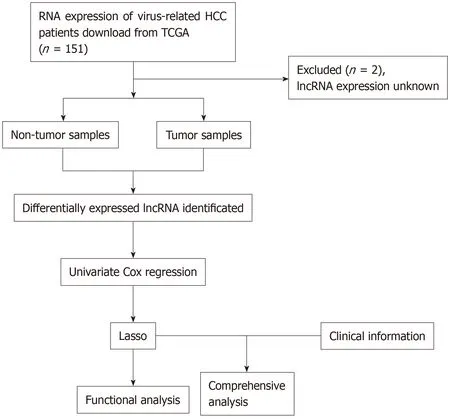
Figure1 Flowchart of study design.HCC:Hepatocellular carcinoma; TCGA:The Cancer Genome Atlas; lncRNA:Long non-coding RNAs.
Functional analysis of lncRNAs for prognosis prediction
Pearson correlation analysis was carried out for the screened lncRNAs as well as the protein coding genes in the TCGA database according to their different expression quantities.Typically,a coefficient of correlation > 0.4 andP< 0.001 would be deemed to show a significant correlation.Enrichment analysis was performed with the DAVID Bioinformatics Tool (version 6.8,https://david-d.ncifcrf.gov/).
RESULTS
DElncRNA identification
The significant DElncRNAs were identified by comparing the tumor samples with non-tumor samples.Altogether,1420 DElncRNAs (including 1358 up-regulated and 62 down-regulated ones) were recognized using the R software “edgeR” package.Afterwards,the heat map (Figure2A) and volcano of top 20 DElncRNAs (Figure2B)were built.
Construction of an lncRNA signature
Univariate Cox regression analysis was performed between DElncRNAs and patients’survival; as suggested by the results,altogether 406 lncRNAs were significantly associated with OS (P< 0.05).Next,LASSO regression was employed for verifying further variables (Figure3).Eight lncRNAs were generated in this process,including AC005722.2,AC107959.3,AL353803.1,AL589182.1,AP000844.2,AP002478.1,FLJ36000,and NPSR1-AS1.Then,the prognostic risk_score was imputed as follows:(0.15103*AC005722.2 expression)+(0.07857*AC107959.3 expression)+(0.09283*AL353803.1 expression)+(0.17744*AL589182.1 expression) + (0.07857*AP000844.2 expression)+(0.09888*AP002478.1 expression)+(0.13460*FLJ36000 expression)+(0.06309*NPSR1-AS1 expression).
Confirmation of the lncRNA signature

Figure2 Heat map and volcano plot.A:Heat map demonstrating the differential expression of long non-coding RNAs (lncRNAs).The X-axis represents the samples,while the Y-axis denotes the differentially expressed lncRNAs.Green color is indicative of the down-regulated genes,while the red color stands for the upregulated genes; B:Volcano plot showing the differential expression of lncRNAs.The X-axis indicates the false discovery rate values after log transformation,whereas the Y-axis suggests the log2 (fold change) values for lncRNA expression.FDR:False discovery rate; FC:Fold change.
The risk score of every case was calculated; then,patients were classified into a highrisk group or low-risk group based on the median threshold (Figure4A and B).The Kaplan-Meier curves for the high-risk as well as low-risk groups are presented in Figure4C.Patients in the high-risk group were associated with a shorter OS compared with those in the low-risk group (P= 2e-10).Meanwhile,the risk score hazard ratio (HR) upon univariate Cox proportional hazards regression was 5.360[95% confidence interval (CI):3.012-9.538] (Table1).Besides,multivariate Cox proportional hazards regression with adjusted clinical covariate also came to the same results (HR=1.94,95%CI:1.61-2.34) (Table2).Subsequently,the ROC curves with time dependency were adopted to assess the prognostic value of the eight-lncRNA signature.In addition,the areas under curves (AUCs) of the prognostic model constructed based on the eight-lncRNA signature were 0.73,0.758,and 0.788 for 1-,3-,and 5-year OS,respectively (Figure4D).
The first node of the conditional inference tree divided the data according to the risk_score,and the lower risk_score (≤0.855) was related to a better OS (P< 0.001)(Figure5).For the lower risk_score (≤0.855),the branch was further divided depending on the stage (P= 0.006),and it was obvious that the risk_score ≤ 0.855 in stage I or II patients was associated with the best survival,while patients with the risk_score > 0.855 had the worst survival.
A nomogram for prognosis was constructed based on the above-mentioned independent factors for prognosis for the sake of visualization as well as prediction,so as to construct an integrated predicting factor for HCC.According to the multivariate Cox regression analysis,the eight-lncRNA risk_score,TNM stage,and race were recognized as independent risk factors (P< 0.05) (Table2).Specifically,the nomogram was constituted by three factors (Figure6A),with a C-index of 0.763(95%CI:0.700-0.826).Moreover,the calibrated curves for the risk of 1-,3-,and 5-year recurrence were highly consistent between the nomogram predicted results and the observed results (Figure6B-D).
Subgroup analysis by etiology
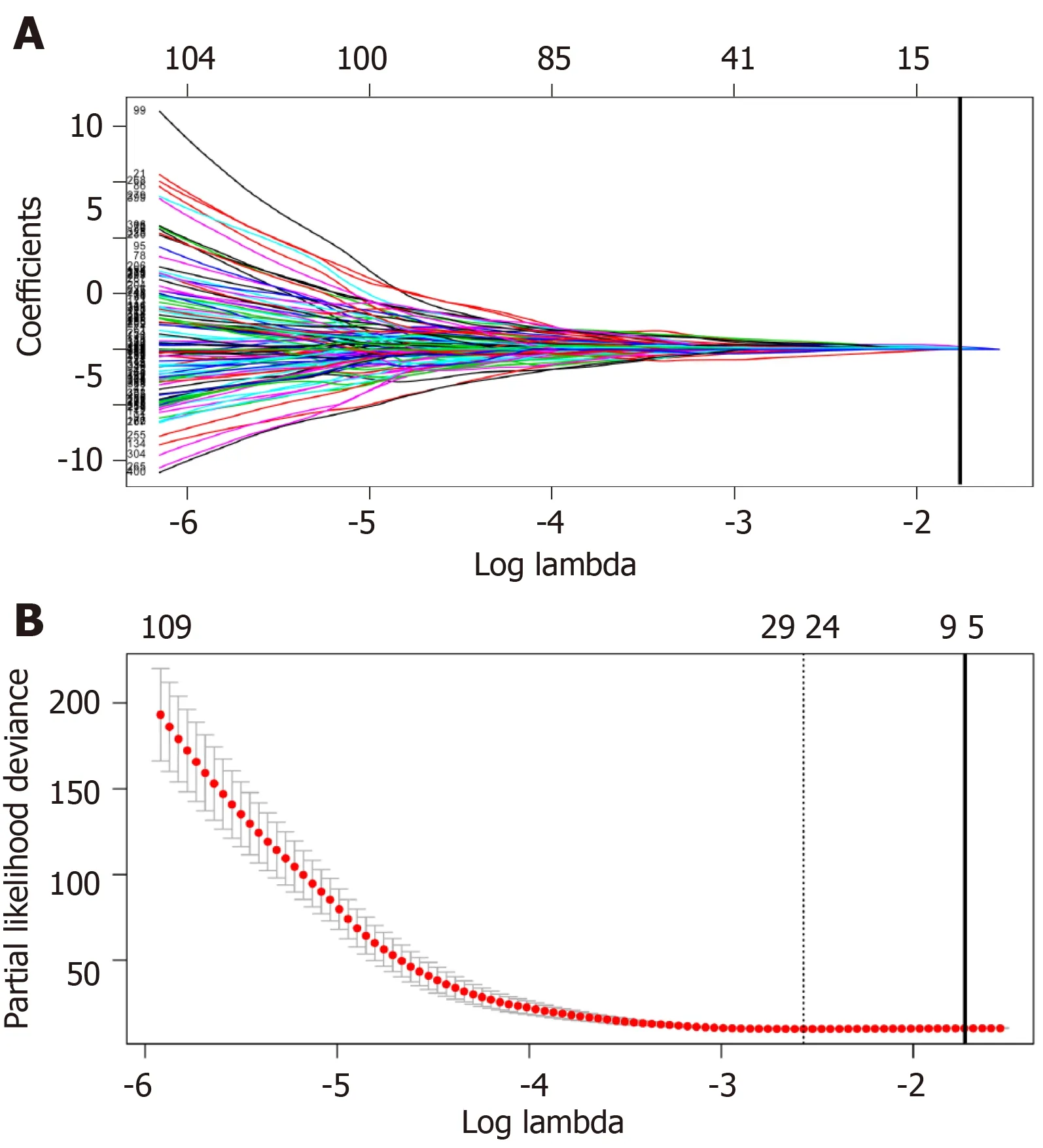
Figure3 Plots of regression coefficient based on least absolute shrinkage and selection operator regression.A:Least absolute shrinkage and selection operator (LASSO) coefficient profiles of the fractions of the 406 significant long non-coding RNAs (lncRNAs) in univariate Cox regression analysis.The solid vertical line implies the optimal lambda value determined by cross-validation; B:Cross-validation for tuning parameter selection in the LASSO model.The vertical lines are drawn at the optimal values by minimum criteria and 1-standard error criteria.And the solid vertical line represents that eight lncRNAs were finally determined.
Subsequently,the patients were further classified into three subgroups,namely,HBV+ HCV (n= 82),HBV alone (n= 58),and HCV alone (n= 9) groups.Kaplan-Meier curves for the high-risk and low-risk groups are displayed in Figure7.The OS of patients benefited from the low-risk score in the HBV + HCV group (P= 6e-5,Figure7A) and HBV alone group (P= 5e-6,Figure7B).At the same time,the AUCs regarding the 1-,3-,and 5-year OS in the HBV + HCV model were 0.681,0.759,and 0.724,respectively (Figure7D),while those in the HBV model were 0.787,0.720,and 0.826,respectively (Figure7E).Curves and correspondingPvalues for HCV patients are also shown in Figure7C and F.
Functional analysis of prognostic lncRNAs
LncRNA-related mRNAs were first filtered out by Pearson correlation analysis and then applied into online tool DAVID for pathway enrichment,so as to examine the possible biological functions of the above-mentioned identified lncRNAs.The top 10 pathways with the highest enrichment are presented in Figure8.Among those,the Wnt signaling pathway,angiogenesis,the p53 pathway,and the PI3 kinase pathway were recognized as the canonical pathways in the initiation as well as progression of HCC.
DISCUSSION
Few HCC prognostic models have been constructed on the basis of lncRNAs up to now.To our knowledge,little is known about lncRNA signature for hepatitis virus infection associated HCC patients.Meanwhile,the majority of studies analyzed virus combined with non-virus related HCC at the same time[11,12].Different from previous studies that carried out analysis regardless of etiology,we conducted the current research in virus positive patients only.The DElncRNAs with marked correlation with OS were comprehensively screened by applying biostatistical methods and univariate Cox analysis.Then,the LASSO regression was applied to analyze the lncRNA data from the carefully chosen patients from TCGA,and eight lncRNAs were identified,which were recognized to be the independent factors for the prognosis of VHCC.Afterwards,the identified significant lncRNAs were employed for constructing a prognostic model,and its prognostic value was verified through Kaplan-Meier analysis,time-dependent ROC curve,and Cox regression analysis.A nomogram was developed to present the constructed risk_score system,and it displayed a favorable accuracy in OS prediction for VHCC.Further functionalanalysis uncovered that several pathways play crucial roles in the effects of the eight lncRNAs on the progression of HCC.
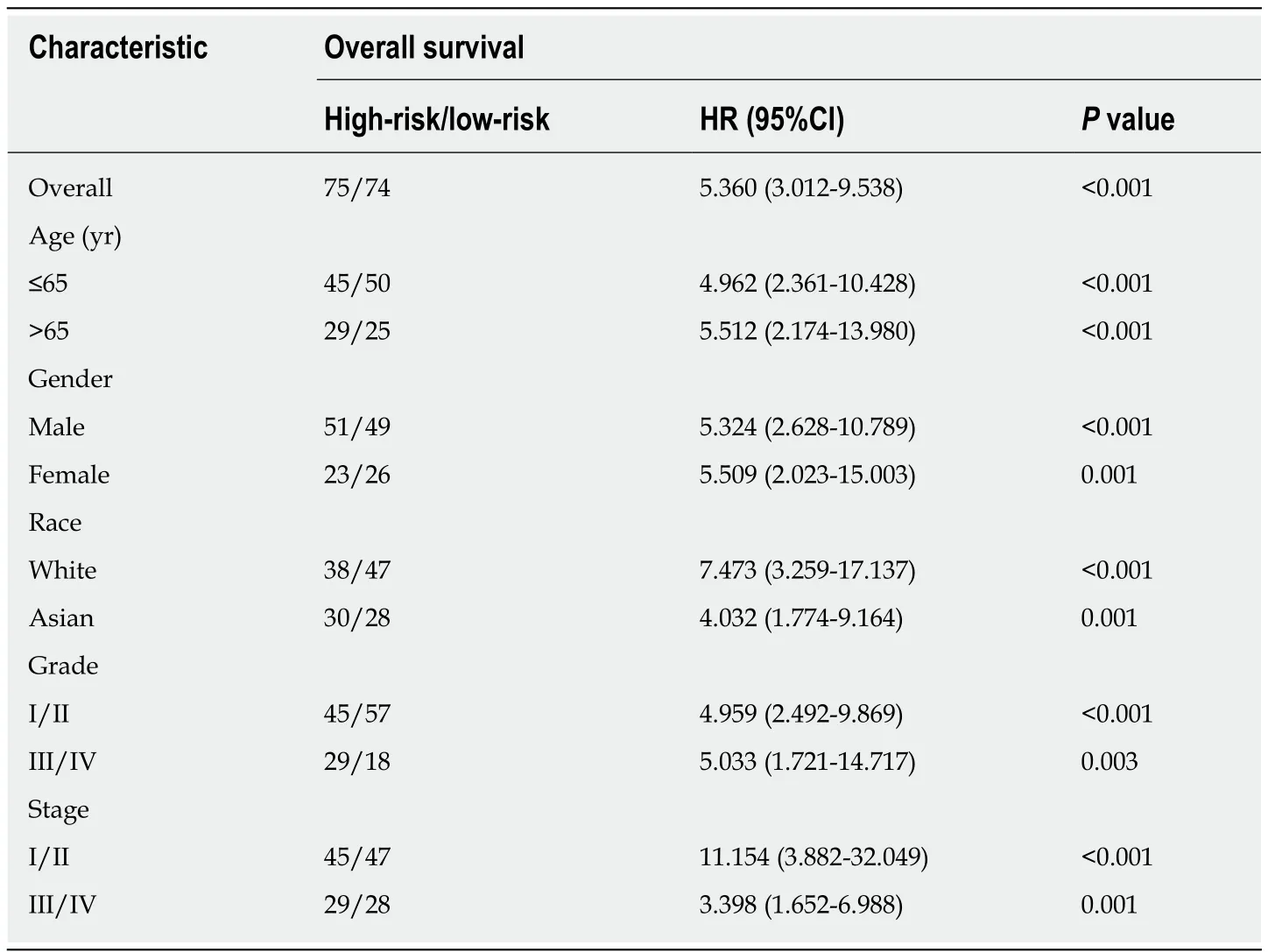
Table1 Stratified overall survival analysis
Our research displayed obvious advantages when compared with other research on the prognostic value of lncRNAs in HCC.First,patient homogeneity was markedly enhanced through careful patient selection before analysis,since the ignorance of patient features might result in false relationships,thus invaliding the conclusions.Existing treatments can treat chronic viral hepatitis,and some research has been performed previously to domenstrate the influence of antiviral treatment on disease progression as well as HCC development[13].Moreover,many studies suggest that the survival of virus-related HCC patients can be prolonged through curative hepatectomy and/or local tumor ablation combined with adjuvant antiviral therapy[14-16].On the other hand,virus and non-virus HCCs have been recognized to have apparently different outcomes,and different therapeutic strategies were recommended for them.Therefore,the prognostic models should also be developed in a distinct way.All the 149 patients were virus positive in our study.Second,with regard to the methodology,the use of LASSO penalized regression could boost the accuracy of bioinformatic analysis.Different from the conventional stepwise regression applied in prior research,the LASSO algorithm contributed to analyzing all independent factors at the same time,which tended to screen the most significant variables[17].Then,the coefficients of the less significant variables would become zero following introduction into the penalty after regularization[18].Consequently,such formulation approach would display a higher accuracy than stepwise regression using the multivariate Cox model,especially in the presence of huge datasets,such as genomics[19].Third,an easy-to-use nomogram model was constructed on the basis of Cox analysis among the carefully screened patients with clinicopathological features,which would facilitate the prediction for individual patients.
As hepatitis viruses vary in different countries[3],our cohort was also divided into three subgroups based on the virus type (namely,HBV alone,HCV alone,and combined).Subgroup survival analysis showed that the as-constructed eight-lncRNA signature performed well in patients with combined virus infection (5-year AUC =0.724),especially for those with HBV infection (5-year AUC = 0.826).No obvious OS heterogeneity was detected among the HCV patients (P= 0.51),which could be resulted from the small sample size (n= 9).
No study had investigated the biological functions for the eight lncRNAs identified in HCC; however,they could affect the genesis as well as the progression of HCC by the Wnt signaling pathway[20-24],angiogenesis[25,26],p53 pathway[27,28],and PI3 kinase pathways[29-31],as suggested by the pathway enrichment results.More importantly,this research could provide new evidence that lncRNAs,whose biological functionshad not been reported before,might potentially serve as the potent markers to predict the prognosis of HCC.However,such findings should be further validated in future studies,and the molecular characteristics should also be investigated.
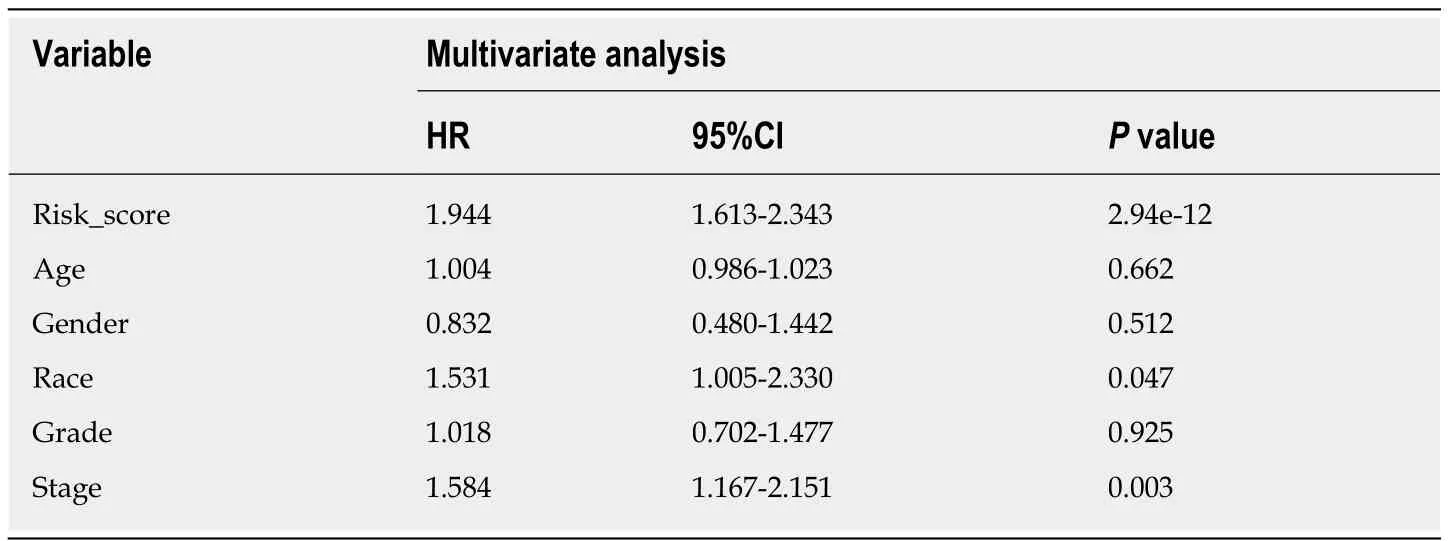
Table2 Cox regression analysis for the overall survival-related clinicopathological features
To our knowledge,this is the first study that constructs an lncRNA nomogram to predict the prognosis of VHCC.Nonetheless,some limitations should be noted in this study.First,no experimental studyin vitroorin vivowas performed to verify the effect of the eight-lncRNA signature,which was constructed based on online datasets by the bioinformatic approach,in predicting the prognosis of HCCs.Second,data regarding more valuable clinical features are not available,including viral hepatitis duration,state of virus,Milan criteria,HCC treatment,and anti-viral treatment.Moreover,our results should be further validated in future studies.
To sum up,a novel lncRNA signature has been uncovered in this study based on comprehensive bioinformatic analysis combined with the expression patterns as well as the clinical information from the carefully selected cohort.Specifically,this risk model can be used as a biomarker to independently predict the prognosis of VHCC.Nonetheless,our results should be validated in studies that investigate the precise mechanisms regarding VHCC progression,as well as those that explore the functions of the eight identified lncRNAs.

Figure4 Establishment and verification of a long non-coding RNA signature for predicting the prognosis of hepatitis virus positive hepatocellular carcinoma using the cut-off value 0.798.A:Distribution of the long non-coding RNA (lncRNA) risk scores; B:Survival status as well as overall survival (OS); C:Kaplan-Meier curve regarding the OS of the high-risk as well as low-risk group classified based on median risk score; D:Receiver operating characteristic curves regarding the 1-,3-,and 5-year survival discriminated by the lncRNA signature.AUC:Area under the curve.

Figure5 Conditional interference tree for prediction.The nodes of the conditional inference tree were calculated automatically based on the influence power of variables.The first node of the tree was risk_score,while the second was stage.Node 3,4,or 5 was the corresponding group a certain patient would be assigned to.For example,a stage I patient with a risk_score ≤ 0.855 would be divided into group node 3,which was related to the best overall survival.
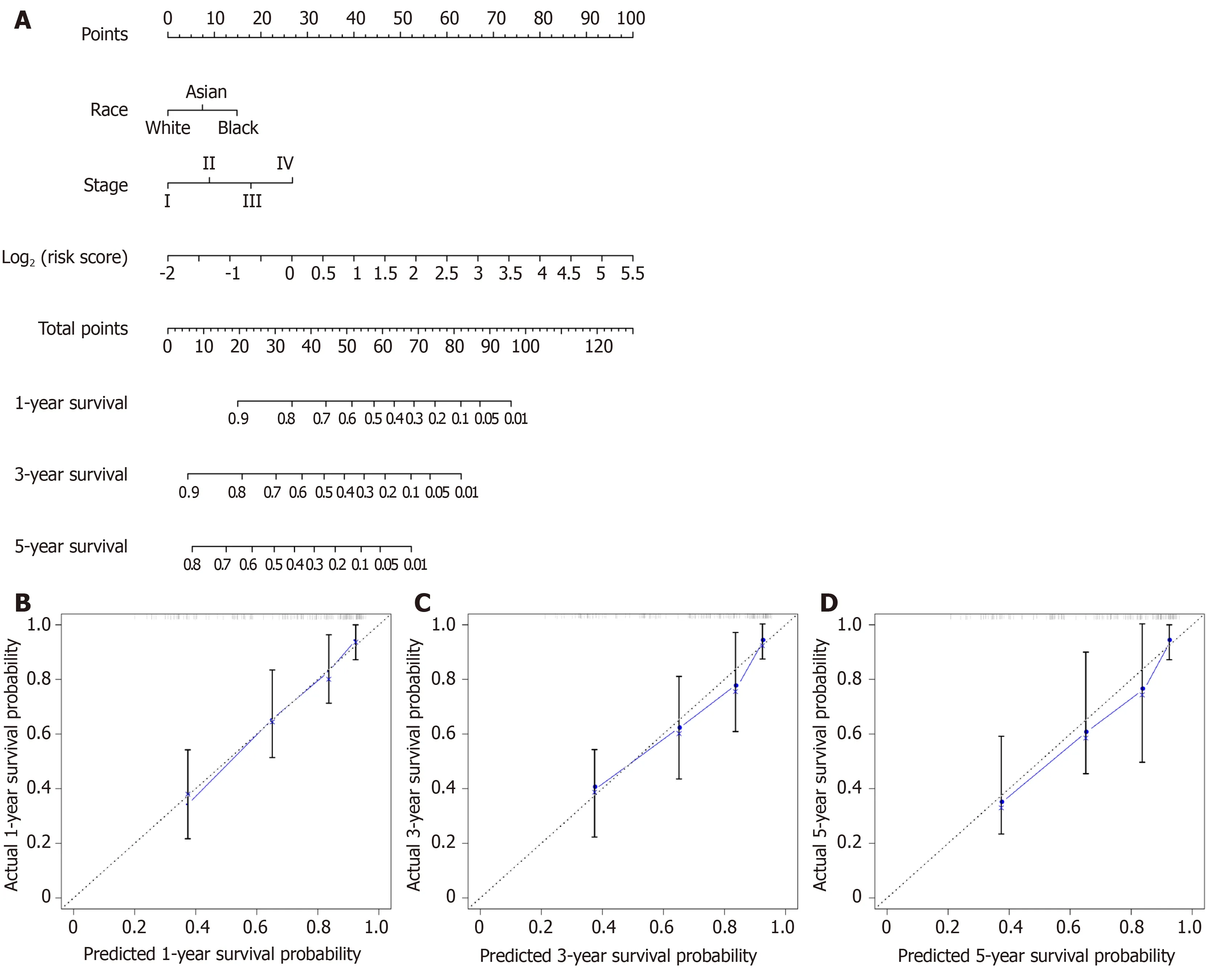
Figure6 Overall survival nomogram predicting 1-,3-,and 5-year survival of hepatocellular carcinoma,and calibrated curves to predict the 1-year,3-years,and 5-year patient survival.A:Overall survival nomogram predicting 1-,3-,and 5-year survival of hepatocellular carcinoma; B:Calibrated curve to predict the 1-year patient survival; C:Calibrated curve to predict the 3-years patient survival; D:Calibrated curve to predict the 5-years patient survival.In calibrated curves,the X-axis represents the survival prediction by nomogram,and the Y-axis represents the actual observation which showed a good agreement with prediction.

Figure7 Subgroup analysis regarding the etiology.A-C:Survival curves of risk_score for hepatitis B virus + hepatitis C virus (HBV + HCV) (A),HBV alone (B),and HCV alone (C) infected patients with HCC; D-F:Receiver operating characteristic curves for HBV + HCV (D),HBV alone (E),and HCV alone (F) infected patients with HCC.HBV:Hepatitis B virus; HCV:Hepatitis C virus.

Figure8 Functional annotations for the eight long non-coding RNAs.The 10 most remarkable pathways enriched based on the co-expressed mRNAs of eight long non-coding RNAs (lncRNAs).Corresponding lncRNAs involved in the deregulated pathways are also presented.A:AC005722.2; B:AC107959.3; C:AL353803.1;D:AL589182.1; E:AP000844.2; F:AP002478.1; G:FLJ36000; H:NPSR1-AS1.
ARTICLE HIGHLIGHTS
Research background
Hepatocellular carcinoma (HCC),one of the most frequent liver cancer subtype,has posed a serious health issue in the world.Hepatitis virus is recognized to be a major factor leading to HCC.Recently,a verity of genetic markers as well as prediction models are proposed to improve HCC treatment.At the same time,numerous statistical techniques are also utilized to mine data in numerous large-scale public databases.Thanks to the well-developed clinical approaches,prognosis models with a higher accuracy and robustness can be established for HCC.
Research motivation
This study aimed to establish a prognosis model on the basis of HCC molecular biomarkers.Typically,long non-coding RNAs (lncRNAs) have been recognized as new predictive factors.Great efforts have been made to establish lncRNA-based HCC models,however,the lncRNA features for hepatitis virus positive HCC (VHCC) are not available at present.
Research objectives
This study aimed to develop a prognostic lncRNA feature for VHCC based on candidate lncRNAs through analyzing data collected from The Cancer Genome Atlas (TCGA) database.
Research methods
Specifically,the least absolute shrinkage and selection operator (LASSO),the most advanced statistic algorithm,was used to establish the prediction model.This approach was carried out on the basis of typical lncRNAs selected according to the expression profiles of lncRNAs collected from the TCGA database.The as-established lncRNA feature was validated,and its clinical applicability was also examined.
Research results
Using LASSO,a risk score system was established to predict the overall survival (OS) for VHCC,which incorporated eight lncRNAs (including AC005722.2,AC107959.3,AL353803.1,AL589182.1,AP000844.2,AP002478.1,FLJ36000,and NPSR1-AS1).Notably,the as-constructed lncRNA feature was helpful in stratifying the risk for VHCC.To extend the model application range,a nomogram was also constructed,which involved both the lncRNA feature and other clinical features.Our results also suggested that the lncRNAs were markedly enriched in the Wnt signaling pathway,angiogenesis,the p53 pathway,and the PI3 kinase pathway.
Research conclusions
The as-constructed signature based on eight lncRNAs displayed a favorable capacity in predicting the prognosis for VHCC patients,which can also contribute to risk stratification and may provide more useful clinical advice for individual patients.
Research perspectives
Our results further support that lncRNAs can serve as possible functional regulating factors for the progression of VHCC.It is the future direction to search for the effective molecular biomarkers and predictive factors for the prognosis of HCC patients.
杂志排行

World Journal of Gastrointestinal Oncology的其它文章
- Efficacy of hybrid minimally invasive esophagectomy vs open esophagectomy for esophageal cancer:A meta-analysis
- Clinical significance of MLH1/MSH2 for stage ll/lll sporadic colorectal cancer
- Endoscopic full-thickness resection for treating small tumors originating from the muscularis propria in the gastric fundus:An improvement in technique over 15 years
- Validation and head-to-head comparison of four models for predicting malignancy of intraductal papillary mucinous neoplasm of the pancreas:A study based on endoscopic ultrasound findings
- Revisiting oral fluoropyrimidine with cetuximab in metastatic colorectal cancer:Real-world data in Chinese population
- Oral chemotherapy for second-line treatment in patients with gemcitabine-refractory advanced pancreatic cancer