基于RAD-seq技术的花红SSR信息分析
2019-12-09冯建文吴亚维罗昌国韩秀梅
宋 莎, 冯建文, 吴亚维, 罗昌国, 韩秀梅
(贵州省农业科学院 果树科学研究所, 贵州 贵阳 550006)
花红(MalusasiaticaNakai.)为蔷薇科(Rosaceae)苹果属(Malus)落叶乔木,果实与苹果相似,果较小,营养价值高,是集花果并赏的树种,我国大部分地区均有分布,在南方可作苹果砧木[1],通过对贵州省苹果属资源调查[2]和亲缘关系分析[3]等发现,花红是贵州省重要的苹果属种质资源。
近年来,多种分子标记被开发,苹果集群分离、全基因组关联分析及功能基因等方面也取得一定进展[4]。巴巧瑞等[5]利用SSR引物和SRAP引物可有效揭示37个品种的遗传多样性;王晓英等[6]利用AFLP分子标记对4个苹果品种进行遗传分析发现,聚类结果与表型特征相符;应用微卫星DNA(SSR)分子标记进行苹果属种质资源鉴定[7-9]和群体遗传分析[10-11]的文献报道也较多。RAD-seq(restriction-site associated DNA sequencing)技术是在二代测序基础上基于全基因组酶切位点的一项简化基因组测序技术,基因组研究热点领域如构建高密度遗传图谱、精确定位重要性状、组装辅助基因组序列、群体基因组学以及系统发生学等应用RAD-seq技术也较多[12]。应用分子标记对贵州花红进行种质鉴定,对保护和利用花红资源具有重大意义。因此,以10份花红资源为材料,利用RAD-seq技术对其进行简化基因组测序,并全面分析花红的SSR信息,以便为花红新型分子标记引物的筛选提供序列参考。
1 材料与方法
1.1 材料
2016年收集花红10份(表1),以实生苗方式保存。2018年从每份材料选取嫩叶5片,液氮速冻,-80℃保存。委托广州基迪奥生物科技有限公司进行RAD-Seq转录组测序(无参)。

表1 供试花红的编号及来源
1.2 DNA提取及质量检测
采用CTAB(十六烷基三甲基溴化铵)法提取材料DNA,NanoDrop微量分光光度计法和1%琼脂糖凝胶电泳法检测DNA质量,以保证其总量和纯度。
1.3 文库构建
利用NEBNextDNA双链片段化酶,根据不同作用时间将dsDNA切割成50~1 000 bp片段。按End Prep 混合酶 3 μL、End Repair Reaction Buffer 6.5 μL、Fragmented DNA 55.5 μL、Total volume 65 μL的反应体系加入试剂进行PCR反应,进行末端修复、磷酸化并加A。按体系Blunt/TA Ligase Master Mix 15 μL、NEBNext Adaptor for Illumina 2.5 μL、Ligation Enhancer 1 μL、Total volume 83.5 μL配制试剂,20℃ PCR反应15 min后,在混合液中加入3 μL USER 酶37℃反应15 min,加上P1接头。带有不同P1接头的样品混合,利用超声或酶切将其打断成300~700 bp的序列,加上P2接头。PCR扩增富集RAD标记。用AMPure XP Beads纯化PCR反应,文库质检。
1.4 信息分析
对每个样品中PE reads的read1进行比对和聚类,得到个体的stacks信息。将多个样本的stacks聚类,得到群体stacks的一致性序列信息。根据群体stacks聚类的结果对read2进行分类,将所有样本来源同一个等位基因位点的read2归为一类。然后对每个等位基因位点的read2进行组装拼接,得到基于reads2拼接的重叠群;将read1聚类得到stacks的一致性序列和read2拼接得到的重叠群进行连接。若两者有重叠,则根据重叠序列进行合并,若两者无重叠,则在中间以N补充。最终得到基于RAD聚类和拼接的RAD标记。以群体RAD 标记为参考序列,重新进行个体的序列比对和变异检测,并进行其他下游高级分析。
利用Treebest 1.9.2构建进化树对样品进行聚类分析。
2 结果与分析
2.1 RAD-seq测序的质量评估
简化基因组测序结果(表2)显示,花红样品的原始数据(Clean date)为1 043 992 564~1 775 387 156个,10个样品共获得13 976 968 360个原始数据;过滤后获得的高质量原始数据占总量的98.18%,Q20为97.31%~97.57%,Q30为93.24%~94.04%,GC含量为38.66%~43.38%,过滤前后的数据差异较小,且Q30处于较高水平,GC含量处于较低水平,表明样本数据量足够,测序数据合格可靠,建库测序成功。
2.2 序列的拼接组装
Reads拼接组装后,重叠群组装总数为628 413,重叠群N50为346 bp,最大长度1 015 bp,最小长度200 bp(图1),平均长度为331 bp。将得到的重叠群继续与reads进行连接构建RAD标记,重叠群组装总数为784 572 bp,重叠群的长度N50=501 bp,最大长度2 210 bp,最小长度156 bp,平均长度为417 bp,可作为后续变异检测和高级分析的参考序列。

表2 RAD-seq基因组测序的花红数据
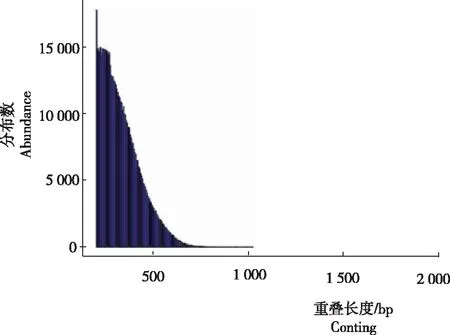
图1Reads组装后长度分布
Fig.1 Contig length of the reads after assembly
2.3 SSR位点及其引物信息
经搜索最终获得SSR位点40 623个,成功设计37 141对SSR引物,成功设计率为91.43%。经SSR检测成功设计引物SSR位点的重复序列长度为16~141 bp,其中最多的是二核苷酸(25 293条),占总数的68.1%;其次是三核苷酸(6 543条),占总数的17.6%;其余依次为四核苷酸(3 380条)、五核苷酸(1 333条)和六核苷酸(592条)。SSR引物基序的重复次数为4~13次,通过梯度退火温度PCR,从10对备选SSR引物中筛选出可在花红中成功扩增并表现出多态性的引物7对,分别为1、2、5、6、8、9、10号,并确定这7对引物的最佳退火温度(表3)。

表3 花红10对引物的特性
2.4 构建系统进化树
由图2可见,10份花红材料可分为两大类,其中贵州的8份材料聚为一类,云南的M-0916和M-0916_2聚为一类。贵州的8份材料中,安龙的M-0708与罗甸的M-0712亲缘关系最近,其余依次为贵阳的JZ、威宁的M-0710、贵阳的M-0706_2、镇宁的M-0702、兴仁的M-0704、兴义的M-0706。

图2 10份花红的系统进化树
3 结论与讨论
种质鉴定的方法有形态学、同工酶、分子标记等。试验采用RAD-seq技术对花红进行简化基因组测序,获得了花红基因组水平上的序列信息,经聚类和组装后也有较高的准确性,可代表花红部分基因组。对获得的序列进行过滤,获得的SSR位点有40 623个,成功设计引物37 141对,从10对备选SSR引物中筛选出可在花红中成功扩增的引物有7对。通过系统进化树对10份花红样品进行聚类分析,可将贵州和云南的材料分开,也能明显看出贵州的8份材料亲缘关系的远近,表明可利用RAD-seq技术进行种质鉴定工作,与异形花[13]和烟草[14]等物种开展的SSR信息研究相类似,RAD-seq技术具有性价比高、数据利用率高、准确性高、不受基因组序列限制等优势[12]。
贵州省属于典型的喀斯特地貌,由于特殊的地理环境,野生果树资源丰富,蕴藏着丰富的基因资源,可能存在值得挖掘的种质。花红是贵州省较常见的资源,但为零星分布,近些年受一些因素的不利影响。贵州省花红资源具有某些独特的基因,与云南省的花红资源易区分,有助于揭示其遗传信息,对未来贵州省花红资源的收集评价及对资源的利用和改良有重要参考价值。但有些花红果实品质良好,经过了较多的人为干预,其具体价值有待进一步研究探讨。
