2-(2-羟基苯亚甲基)胺基-3-氰基吡啶的结构和光谱性质
2019-12-06孟德素卢金凤武利顺庞艳玲张丽莹
孟德素,卢金凤,武利顺,庞艳玲,张丽莹
(菏泽学院 化学化工学院,菏泽 274015)
1 引 言
Schiff碱及其络合物一直是合成研究的热点,由于其具有很好的非线性光学性[1]、光致变色性[2]和导电性[3]等特殊性, 在信息存储、光开关以及信息显示等领域存在潜在应用价值,因而受到了人们的关注.Schiff碱由于具有特殊的荧光基团的特性,可以作为探测某种金属离子的荧光探针[4],也可以在电致发光器件上得到较纯的荧光,使其在有机电致发光器件上得到了广泛应用[5].另外Schiff碱及其配合物在功能材料、催化剂及超分子设计等领域有广泛的应用[6,7].目前,对Schiff碱的研究主要是从实验方面进行探讨,而从理论角度进行的研究相对较少,所以对Schiff碱类化合物进行量子化学研究,有助于了解其结构与光谱活性,为有目的设计合成Schiff碱化合物奠定一定的基础.目前常用的量子化学方法中的密度泛函理论被广泛的应用于分子电子结构和光谱性质的理论研究中,计算分子结构和振动光谱等参数,并与实验数据符合的较好[8].
本文实验合成了Schiff碱 2-(2-羟基苯亚甲基)胺基-3-氰基吡啶,并对其进行了红外光谱和拉曼光谱检测,利用密度泛函理论B3LYP方法在6-311++G**基组上优化了分子的几何构型,计算了分子的振动光谱数据及强度,通过实验和理论数据的结合,对振动光谱进行了指认,并分析了其前线分子轨道,得到预测Schiff碱类化合物结构和振动光谱的理论方法,为进一步研究Schiff碱类化合物提供了基础.
合成路线如下:
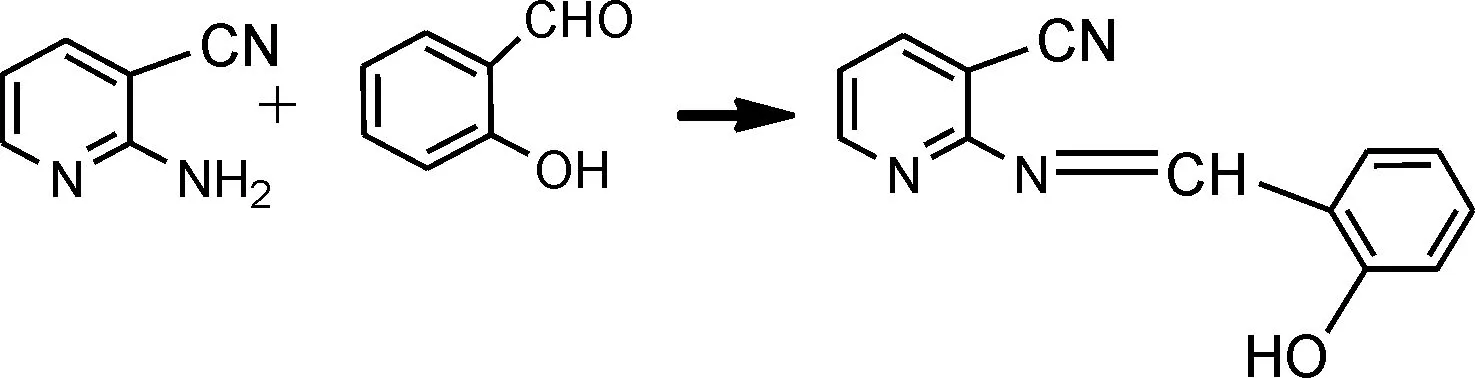
2 实 验
2.1 仪器与试剂
AVATAR-370傅里叶红外光谱仪(美国尼高利公司);MP-21型显微熔点仪(上海精密仪器仪表有限公司);NEXUS-670激光拉曼光谱仪(美国尼高利公司).
水杨醛(武汉有机实业股份有限公司);2-氨基-3-氰基吡啶(湖北巨胜科技有限公司);无水乙醇(天津市科密欧化学试剂有限公司).
2.2 Schiff碱2-(2-羟基苯亚甲基)胺基-3-氰基吡啶的合成
在50 mL圆底烧瓶中加入0.721 g(6 mmol)2-氨基-3-氰基吡啶、0.732 g(6 mmol)水杨醛,10 mL无水乙醇,加热搅拌回流2.5 h,冷却,减压蒸馏,冷却后析出黄色固体,抽滤,干燥,用无水乙醇进行重结晶,得到Schiff碱2-(2-羟基苯亚甲基)胺基-3-氰基吡啶1.23 g,黄色晶体,产率88.4 %.
2.3 光谱检测
在室温下用AVATAR-370傅里叶红外光谱仪,KBr压片,波数范围在4000-400 cm-1,分辨率为2 cm-1,测定Schiff碱的红外光谱.用NEXUS-670激光拉曼光谱仪,波数范围在3800-50 cm-1,分辨率为0.1 cm-1,测定Schiff碱的拉曼光谱.
2.4 量子化学计算方法
利用B3LYP/6-311++G**理论对Schiff碱2-(2-羟基苯亚甲基)胺基-3-氰基吡啶分子进行结构优化,得到了其键长、键角和二面角等结构参数,同时在优化结构的基础上计算了该分子的红外光谱和拉曼光谱,并对振动模式进行了全面的指认和分析.
3 结果与讨论
3.1 分子的几何构型
用B3LYP/6-311++G**理论对Schiff碱2-(2-羟基苯亚甲基)胺基-3-氰基吡啶分子进行了结构优化,得到两种能量较低的构象列于图1中,优化的结构参数列于表1中.在优化出的构象里,b构象能量为-740.45169231 a.u.比a构象能量-740.43820143 a.u.低,能量越低结构越稳定.所以,构象b为Schiff碱分子最稳定的结构.以下均是对Schiff碱稳定构象b的讨论.
从表1中的二面角数据可以看出:除了C(2)-C(1)-N(12)-C(13)和N(6)-C(1)-N(12)-C(13)两个二面角分别是159.729 °和-22.054 °以外,其他二面角的绝对值均近似180 °或0 °,表明苯环和吡啶环之间存在着绝对值为22 °的二面角,C(15)-C(16)-C(17)-C(18)-C(19)-C(20)苯环与羟基和-CH=N-在同一平面上,形成了大的共轭体系.从键长的数据发现:schiff碱分子中苯环的C(17)-C(19)的键长为1.386 Å,C(18)-C(20)的键长为1.390 Å,都比单个苯环中C-C键的键长1.402 Å短,而C(15)-C(16)的键长为1.410 Å,C(15)-C(17)的键长为1.406 Å,都比单个苯环中的C-C键要长;而C(13)-C(15)的键长为1.459 Å,比正常的中C-C键的键长1.540 Å要短,而N(12)-C(13)的键长为1.285 Å比正常的C=N键的键长1.270 Å要长,C(16)-O(25)的键长为1.365 Å比正常的C-O键的键长1.430 Å要短.从这些数据中可以看出Schiff碱分子双键键长变长,单键键长变短,说明分子中存在大的共轭体系.
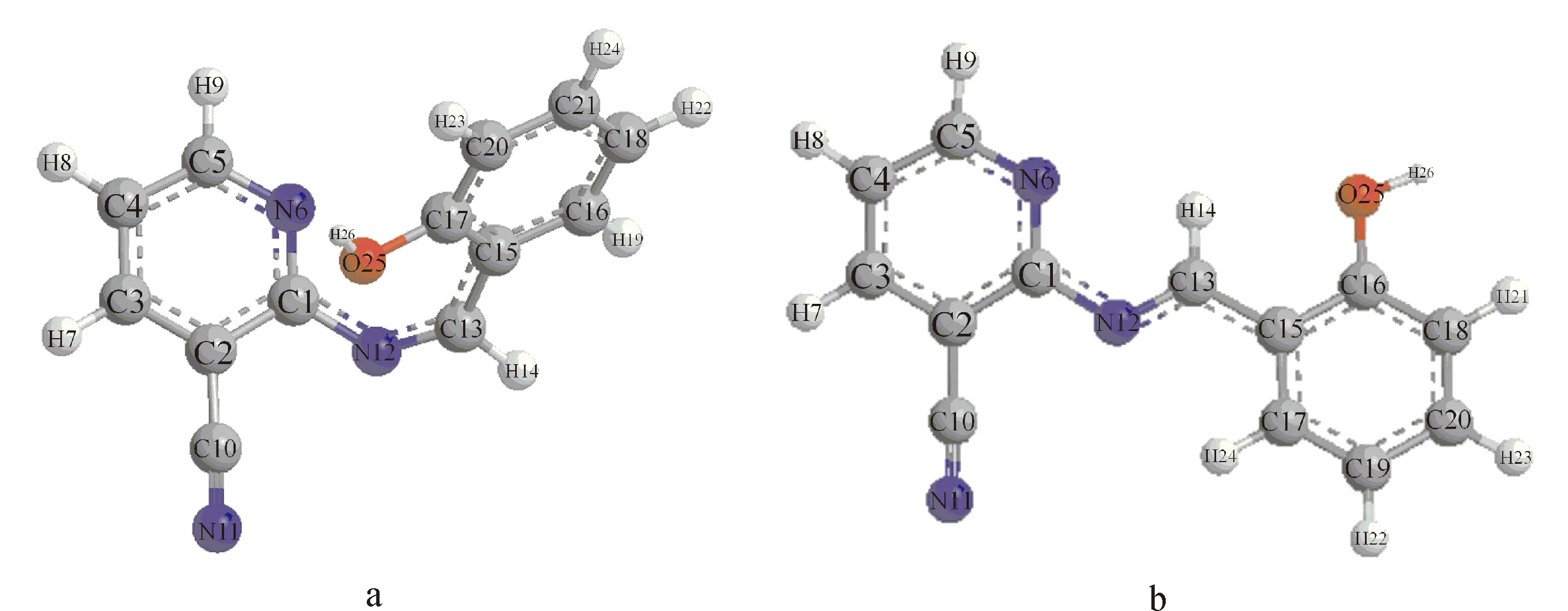
图1 用B3LYP/6-311++G**理论优化出的Schiff碱的优势构象a和bFig.1 Optimized molecular structures a and b of Schiff base at B3LYP/6-311++G** level.

键长计算值(Å)键角计算值(°)二面角计算值(°)O(25)-C(16)-C(18)-C(20)-179.881O(25)-C(16)-C(18)-H(21)0.062
3.2 振动光谱分析
按照2.3实验中的方法测定Schiff碱的傅里叶变换红外光谱和傅里叶拉曼光谱,按照2.4的计算方法得到理论的红外和拉曼光谱的振动频率及其强度,然后将实验光谱图和计算光谱图做比较,见图2和图3.

图 2 Schiff碱的红外光谱图:(a)实验测定(b)理论计算Fig. 2 infrared spectrum of Schiff base:(a)experimental determination(b)theoretical calculation
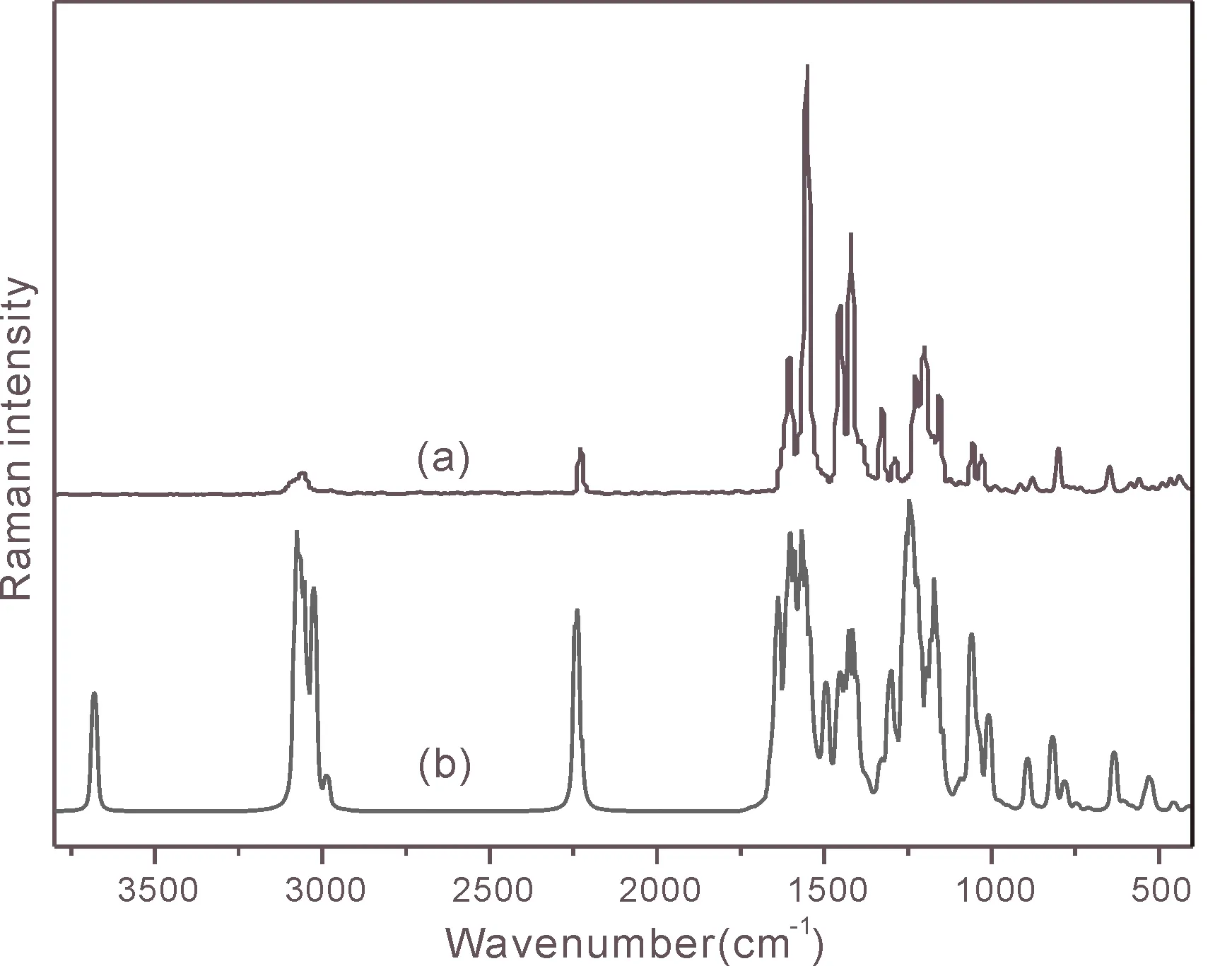
图 3 Schiff碱的拉曼光谱图:(a)实验测定(b)理论计算Fig. 3 Raman spectrum of Schiff base:(a)experimental determination(b)theoretical calculation
通过对比图2和图3,发现理论计算和实验得到的谱图对应的特征峰是相吻合的,但个别峰的位置不同,这可能是由于理论计算时只考虑到单个分子,没有考虑分子间的作用力,而实验测定的分子是多个分子的堆积,分子间具有相互作用.
该Schiff碱属于C1点群,有72 个简正振动模式.在优化Schiff碱结构的基础上进行了振动频率分析,给出了红外光谱和拉曼光谱,为实验上各种吸收峰的归属提供了帮助.考虑到B3LYP的系统误差,引入了矫正因子,当频率小于1700 cm-1使用矫正因子0.98,频率1700 cm-1以上使用矫正因子0.96[9].表2列出了分子的矫正前与矫正后的计算频率、强度,实验测定的频率及对应的振动模式的归属.
3.2.1O-H的振动
在红外光谱中的O-H伸缩振动谱带强,游离的醇和酚的 νO-H在3700 ~ 3500 cm-1[10]以内峰形尖而强,缔和的羟基在3500-3200 cm-1[11]以内峰形强而宽.对于Schiff碱分子的O-H伸缩振动频率,对应红外实验测定值是3437.07 cm-1,而理论计算值在3681.35 cm-1处.这是由于在实验中OH具有很强的极性,可以和其他分子产生缔合,引起频率的较大位移,而在拉曼光谱中没有明显的吸收.O-H的面内弯曲振动吸收带在1500~1300 cm-1[12]附近,理论计算值在1330.76 cm-1处,对应实验测定值是1350.44 cm-1,而拉曼光谱中实验测定值为1328.8 cm-1.
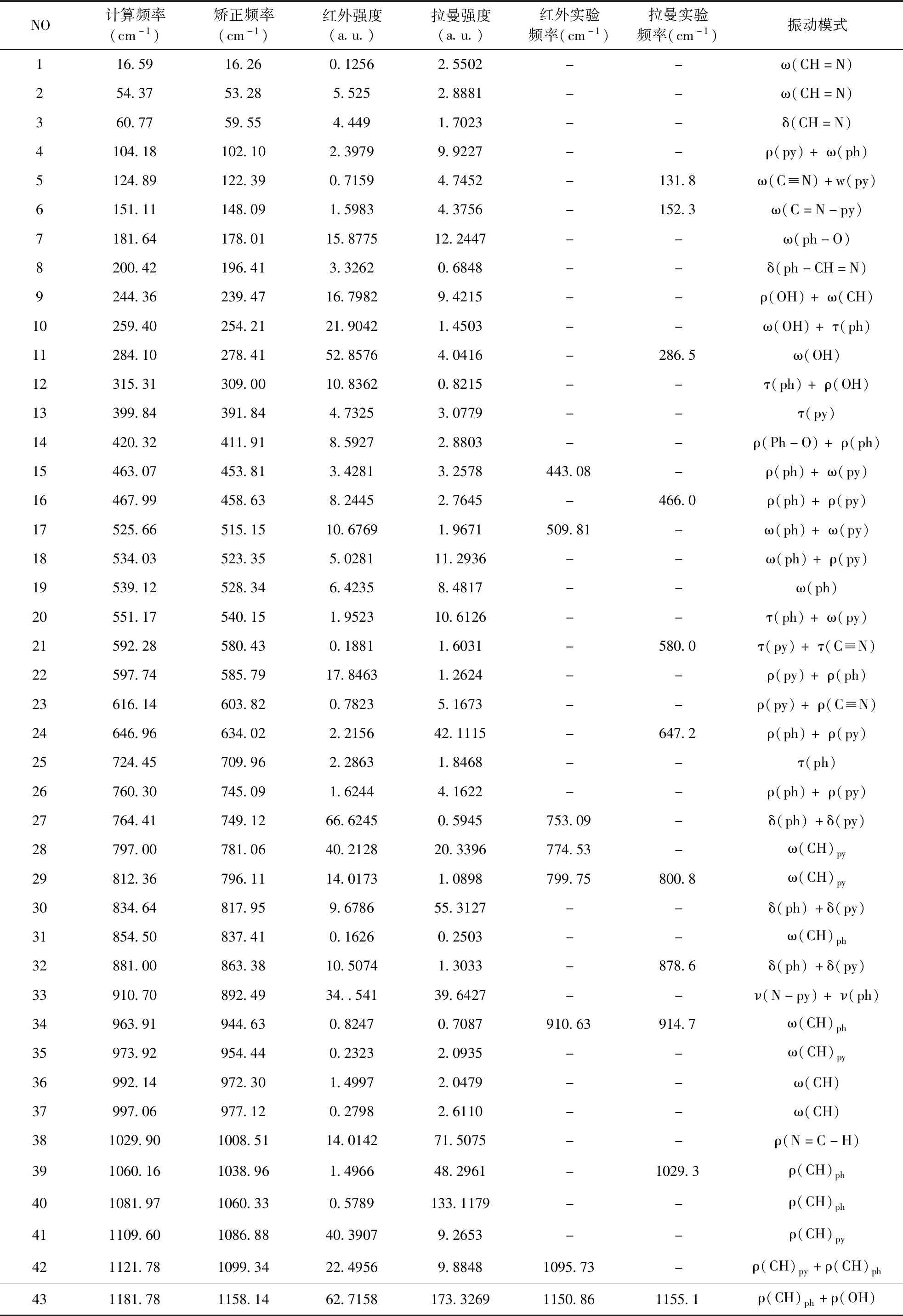
表 2 Schiff碱的计算频率、实测频率、强度及对应的振动模式
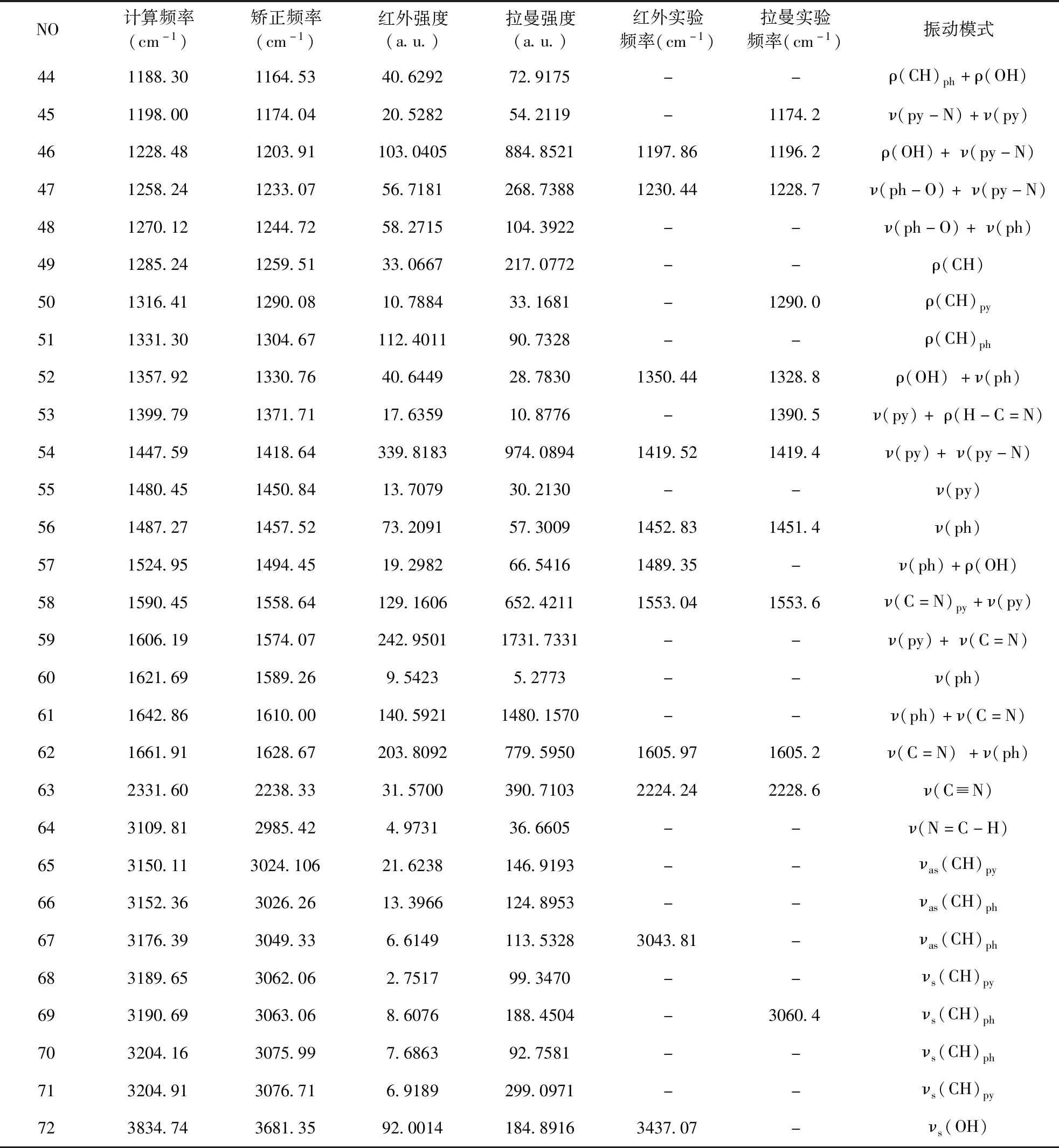
NO计算频率(cm-1)矫正频率(cm-1)红外强度(a.u.)拉曼强度(a.u.)红外实验频率(cm-1)拉曼实验频率(cm-1)振动模式441188.301164.5340.629272.9175--ρ(CH)ph+ρ(OH)451198.001174.0420.528254.2119-1174.2ν(py-N)+ν(py)461228.481203.91103.0405884.85211197.861196.2ρ(OH)+ν(py-N)471258.241233.0756.7181268.73881230.441228.7ν(ph-O)+ν(py-N)481270.121244.7258.2715104.3922--ν(ph-O)+ν(ph)491285.241259.5133.0667217.0772--ρ(CH)501316.411290.0810.788433.1681-1290.0ρ(CH)py511331.301304.67112.401190.7328--ρ(CH)ph521357.921330.7640.644928.78301350.441328.8ρ(OH)+ν(ph)531399.791371.7117.635910.8776-1390.5ν(py)+ρ(H-C=N)541447.591418.64339.8183974.08941419.521419.4ν(py)+ν(py-N)551480.451450.8413.707930.2130--ν(py)561487.271457.5273.209157.30091452.831451.4ν(ph)571524.951494.4519.298266.54161489.35-ν(ph)+ρ(OH)581590.451558.64129.1606652.42111553.041553.6ν(C=N)py+ν(py)591606.191574.07242.95011731.7331--ν(py)+ν(C=N)601621.691589.269.54235.2773--ν(ph)611642.861610.00140.59211480.1570--ν(ph)+ν(C=N)621661.911628.67203.8092779.59501605.971605.2ν(C=N)+ν(ph)632331.602238.3331.5700390.71032224.242228.6ν(C≡N)643109.812985.424.973136.6605--ν(N=C-H)653150.113024.10621.6238146.9193--νas(CH)py663152.363026.2613.3966124.8953--νas(CH)ph673176.393049.336.6149113.53283043.81-νas(CH)ph683189.653062.062.751799.3470--νs(CH)py693190.693063.068.6076188.4504-3060.4νs(CH)ph703204.163075.997.686392.7581--νs(CH)ph713204.913076.716.9189299.0971--νs(CH)py723834.743681.3592.0014184.89163437.07-νs(OH)
注:ν-伸缩振动;ω-面外摇摆振动;ρ-面内摇摆振动;δ-面内弯曲振动;τ-扭曲振动;s-对称;as-不对称
Ph: C15-C16-C17-C18-C19-C20形成的苯环 py: C1-C2-C3-C4-C5-N6形成的吡啶环
3.2.2C-H的振动
从图3和表2可以看出,3060.4 cm-1处有明显的拉曼峰,与理论计算值3063.06 cm-1相符,归属于苯环上C-H的对称伸缩振动,3043.81 cm-1处的红外峰与理论计算中得到的3049.33 cm-1值对应于苯环上C-H的不对称伸缩振动,吡啶环上C-H伸缩振动在红外和拉曼实验中没有明显的吸收.实验中红外光谱位于910.63 cm-1处的峰和拉曼光谱位于914.7 cm-1处的峰,归属为苯环C-H面外摇摆振动,由实验测得的红外频率为774.53、799.75 cm-1和拉曼频率为800.00 cm-1的峰指认为吡啶环C-H面外摇摆振动,与文献[12]中给出的结果(C-H面外摇摆振动频率在960~700 cm-1)相一致,也与理论计算值吻合.芳环上C-H面内摇摆振动频率一般在1300 ~ 1000 cm-1区域[12],实验中拉曼光谱位于1029.3、1151.1和1290.0 cm-1三处的峰与红外光谱位于1095.73、1150.86 cm-1两处的峰,归属为芳环C-H的面内摇摆振动.
3.2.3C≡N、C=N和C-O的振动
有机物中C≡N的特征振动在2600~2200 cm-1区域[13],红外实验值2224.24 cm-1和拉曼测定值2228.6 cm-1归属于C≡N的伸缩振动,与理论计算得到的2284.96 cm-1峰基本相符合.红外实验测得的l605.97 cm-1峰与拉曼测定的l605.2 cm-1峰归属为非环状共轭的C13=N12的伸缩振动,对于环共轭的C=N伸缩振动,不但考虑环共轭体系的C=N吸收,还要考虑它与其他双键相互作用,C1=N6和C5=N6的伸缩振动的红外实验测定值为1553.04 cm-1,拉曼实验值为1553.6 cm-1,其中在很大程度上耦合了吡啶环的振动,其理论值对应为1558.64 cm-1.C-O的伸缩振动在酚和醇中位于1250~1050 cm-1区域[14],Schiff碱的C-O伸缩振动在红外实验中是1230.44 cm-1,拉曼实验中是1228.7 cm-1,与理论计算值1233.07 cm-1相对应.
3.2.4苯环和吡啶环的振动
芳烃C-C伸缩振动通常在1650~1200 cm-1处[15],实验测定Schiff碱分子的红外光谱在1489.35,l452.83 cm-1处的峰和拉曼光谱在l451.4 cm-1处的峰归属为苯环的C-C伸缩振动,与理论计算值1494.45,l457.52,1330.76 cm-1相符.计算得到的1558.64,1418.64,1371.71 cm-1峰归属于吡啶环的振动,与文献[12]中给出的结果(吡啶环的振动频率在1650~1350 cm-1之间)相一致,对应于红外实验值1419.52 cm-1,拉曼实验值为1419. 4,1390.5 cm-1.芳环的面内弯曲振动在650~900 cm-1,计算得到的863.38,749.12 cm-1峰归属为吡啶环及苯环的弯曲振动,与红外实验测量结果753.09 cm-1和拉曼光谱测定值878.6 cm-1相一致.
通过以上实验光谱与理论数据比较发现,理论计算的频率值与实验结果吻合的较好.但个别振动频率的计算值和实验值有一定的差别,原因在于理论计算时只考虑到单个分子,没有考虑分子间的作用力,而实验测定分子时,分子间具有相互作用;另外,计算中得到的大多数振动频率都由多个振动模式叠加而成的,也会使理论计算值与实验数据有些不符.对拉曼光谱的分析可以看出C=N亚胺键部分和苯环的振动比较强,说明C=N亚胺键的电子容易极化,为金属离子络合形成配位化合物提供了有利条件.在计算振动频率方面,说明B3LYP方法在研究相关的Schiff碱体系方面,具有较好的应用性.
3.3 前线分子轨道分析
最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)称为前线分子轨道.前线分子轨道决定分子的重要性质如电、光、紫外可见光谱及化学反应[16].图4为Schiff碱在B3LYP/6-311++G**理论水平计算得到的前线分子轨道的能量分布图.HOMO能量为-0.24154 a.u.,LUMO能量为-0.09207 a.u.,可见最高占据轨道能量较低,最高占据轨道与最低未占轨道的能隙差ΔE=0.14947 a.u,差值较小,分子不稳定,易参与化学反应.
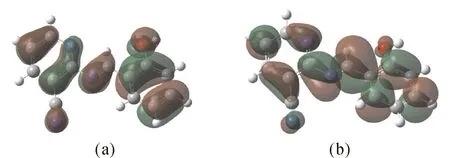
图 4 Schiff碱的前线分子轨道图(a)HOMO; (b) LUMOFig. 4 The frontier molecular orbits of Schiff base(a)HOMO; (b) LUMO
从图4可以看出,最高占据轨道主要集中在苯环及相连的C=N亚胺键部分,最低未占用轨道主要集中在苯环、吡啶环和C=N亚胺键上,这说明苯环和C=N亚胺键具有接受电子和提供电子的能力.以苯环和吡啶环这两个做对比可以发现,无论是HOMO或LUMO,苯环占有的比例都比吡啶大,这是因为苯环与亚胺键连接,形成了共轭体系,这与前面的键长分析结果是一致的.由HOMO轨道图可以看出O与C=N亚胺容易提供电子,可以与金属离子配位,形成配合物.
4 结 论
实验合成了Schiff碱2-(2-羟基苯亚甲基)胺基-3-氰基吡啶,结合密度泛函理论和两种光谱实验方法得到了Schiff碱的拉曼光谱和红外光谱,在6-311++G**基组水平上,对化合物的结构进行优化,并对主要官能团的振动模式进行归属,通过实验和理论的对比,结果表明理论计算结果和实验数据基本相吻合.另外通过对分子轨道的分析说明Schiff碱的HOMO与LUMO的能隙差ΔE=0.14947 a.u.,差值较小,易参与化学反应,分子中C=N亚胺容易提供电子,可以与金属离子配位,为Schiff碱类化合物的进一步研究提供了理论基础.
