H2O2在α-Fe2O3表面非均相氧化SO2机理的模拟研究
2019-11-28李冬坤董发勤李海龙霍婷婷贺小春赵玉连
李冬坤,董发勤,李海龙,霍婷婷,贺小春,赵玉连,彭 洁
(1. 西南科技大学 环境与资源学院, 四川 绵阳 621010; 2. 固体废物处理与资源化教育部重点实验室,四川 绵阳 621010; 3. 西南科技大学 材料科学与工程学院, 四川 绵阳 621010)

过氧化氢(H2O2)是一种重要的大气氧化剂,与对流层中HOx自由基收支和氧化能力直接相关。在实际大气中,矿物气溶胶上的非均相反应是SO2、气态H2O2重要的汇(Reeves and Penkett, 2003; Pradhanetal., 2010; Zhaoetal., 2013)。Song和Zhao等结合计算模拟和实验研究证实了H2O2能够在过渡金属氧化物、矿物粉尘表面分解产生·OH自由基(Zhaoetal., 2014; Songetal., 2017, 2018)。·OH自由基的存在更有利于矿物气溶胶上的非均相氧化的发生(Bouya, 2015)。此外,Huang等发现H2O2的存在可以提高SO2在矿物粉尘上的吸附率——在有H2O2存在的情况下,SO2的饱和覆盖率比无H2O2存在的情况下大5倍左右。另外,在有H2O2存在的干燥条件下,真实矿尘对SO2的吸收系数约提高30%~50%(Huangetal., 2015, 2016)。然而,目前关于H2O2的这种促进作用及H2O2在过渡金属氧化物α-Fe2O3上氧化SO2的非均相反应机理信息仍知之甚少,SO2和H2O2共存时在α-Fe2O3上的优先吸附顺序以及SO2是如何与矿物氧化物结合并发生转化的途径仍不够清楚,需要做进一步的研究。
因此,本文选取大气中无机气体(SO2)、H2O2和赤铁矿(α-Fe2O3)作为研究对象,采用巨正则蒙特卡罗方法(Grand Canonical Monte Carlo, GCMC)和密度泛函理论,模拟计算了SO2、H2O2在α-Fe2O3(001)表面不同吸附构型的吸附和非均相氧化作用,进一步分析SO2、H2O2在α-Fe2O3上的非均相氧化机制,为更好地评估SO2、H2O2非均相反应对大气中矿物氧化物老化作用提供重要信息。
1 计算细节
1.1 α-Fe2O3晶体结构模型建立
选用具有最稳定结构的铁氧化物赤铁矿(α-Fe2O3)为对象,建立α-Fe2O3晶体结构模型。该构型主要基于氧原子的六方密排晶格,具有R-3C立方空间群结构。其中Fe原子仅仅占据了氧原子之间的2/3的氧八面体空位,如图1所示。经过优化后的结构参数(a=b=5.03 Å,c=13.77 Å;α=β= 90°,γ=120°)与实验报道的结构参数(a=b=5.04 Å,c=13.75 Å;α=β=90°,γ=120°)较一致(Songetal., 2013)。Weiss和Song等人的理论计算研究发现包含Fe—O3—Fe端面的α-Fe2O3(001)晶面是主要的自然生长晶面,其催化活性高于其它晶面(Weissetal., 2002; Songetal., 2017),因此本文主要讨论α-Fe2O3(001)晶面与SO2间的非均相反应。为了研究α-Fe2O3与SO2的相互作用,所有结构模型均采用了包含4个α-Fe2O3原胞结构的周期性重复的超晶胞(2×2×1),且在z轴方向建立了16 Å的真空层,并考虑了α-Fe2O3(001)表面上不同的吸附位点(Fe位和O位)和吸附分子不同的吸附取向对吸附机制的影响。
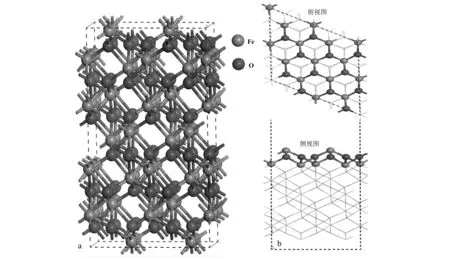
图 1 α-Fe2O3晶体结构图(a)和α-Fe2O3(001)表面的俯视图和侧视图(b)Fig. 1 Crystal structure diagram of α-Fe2O3 (a), top and side views of the surface of α-Fe2O3 (001) (b)
1.2 计算方法
本文中涉及的计算均借助于美国Accelrys公司提供的Material Studio软件。首先,采用GCMC方法将结构优化后的SO2、H2O2分子吸附在α-Fe2O3(001)表面,通过考虑不同的吸附位点(Fe位和O位),建立不同的吸附结构模型并对吸附体系的相互作用能进行计算。其次,在不同的系宗(NVE和NPT)中采用共轭梯度的方法,利用Castep模块中的Geometry Optimization和Dynamic对不同的吸附结构模型进行结构优化和分子动力学驰豫,保证不同的吸附结构模型达到平衡态。其中,短程的范德华力和长程的静电相互作用分别用Atom-Based和Ewald+Group方法进行模拟,温度和压力分别由Nose-Hoover thermostat和Berendsen-Barostat方法控制。最后,在相同的计算模块(Castep模块)中,采用基于广义梯度近似(Generalized Gradient Approximation,GGA)的PBE(Perdew-Burke-Ernzerhof)交换关联泛函和投影缀加平面波法(Augmented-Plane-Wave,APW)计算了体系的电子结构。在考虑强关联体系中的高度局域化Fe-3d轨道作用的前提下,本文采用超软赝势(Ultrasoft Pseudopotentials)和GGA+U的方法计算Fe-3d轨道能级结构(Bianetal.,2017)。其中,Castep模块参数为: Monkhorst-Pack的布里渊区为3×3×3,GGA+PBE描述电子间的交换关联函数,超软赝势描述离子实与价电子之间的相互作用。自洽场运算中采用Pulay密度混合法,平面波截止能为300 eV,收敛精度SCF为2×10-5eV/atom。对于H2O2和SO2在α-Fe2O3(001)表面上的吸附能计算依赖于公式:
E=ET- (EF+Es)
式中的ET指H2O2或者SO2吸附在α-Fe2O3表面上的总能,EF指孤立的H2O2或者SO2分子的总能,ES指独立的α-Fe2O3(001)表面的总能。负的吸附能表明H2O2或者SO2分子在α-Fe2O3(001)表面的吸附是放热反应,负的吸附能的绝对值越大,表明分子与表面的相互作用越强。
2 结果与讨论
2.1 SO2在α-Fe2O3 (001)表面的吸附
SO2表面吸附是SO2在α-Fe2O3(001)上发生非均相反应的重要步骤。考虑到不同的吸附位点,对SO2在α-Fe2O3(001)表面上不同吸附位点吸附的结构进行了优化,得到两种稳定的吸附构型1A(Fe—S)和1B(Fe—O),如图2所示。其中,1A和1B构型是由SO2分别通过S原子和O原子与表面的Fe原子进行吸附,相应吸附构型1A的结构参数为: Fe—S=2.43 Å,Fe—O=1.73 Å;吸附构型1B的结构参数为: Fe—O=1.93 Å; S—O=1.72 Å。吸附能如表1所示。
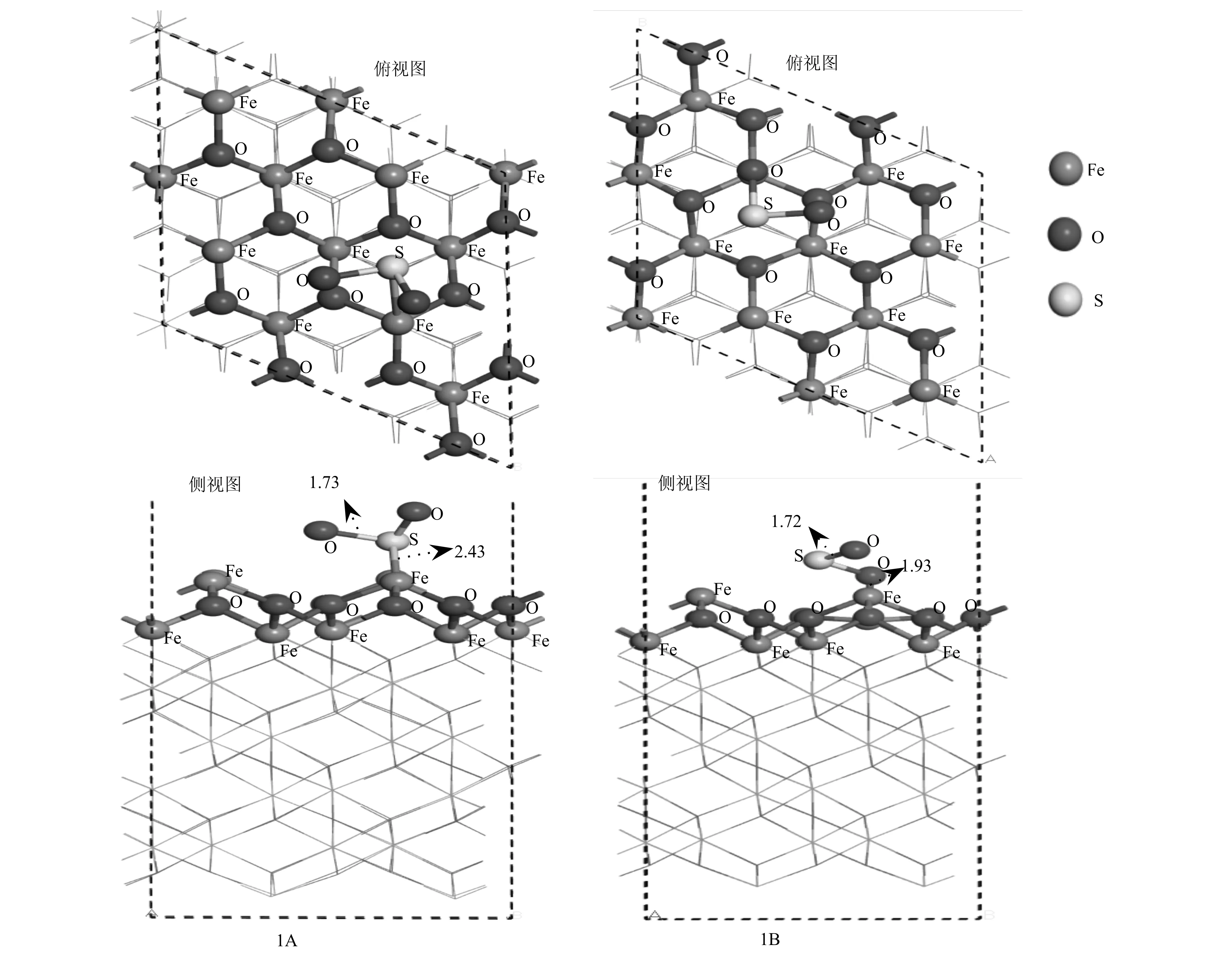
图 2 SO2在α-Fe2O3(001)表面的两种吸附构型Fig. 2 Two adsorption configurations of SO2 on the surface of α-Fe2O3 (001)
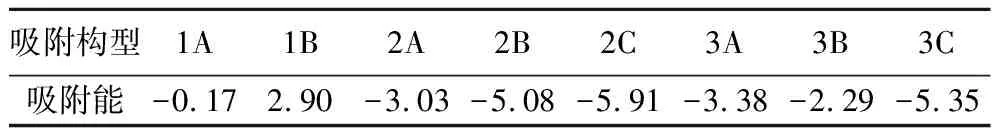
表 1 不同吸附构型的吸附能 E/eVTable 1 Adsorption energy of different adsorption configurations
由表1可以看出,吸附构型1A的吸附能(-0.17 eV)明显小于吸附构型1B的吸附能(2.90 eV),由此说明SO2在α-Fe2O3(001)表面的化学吸附是通过SO2的S原子和α-Fe2O3的Fe原子发生的。SO2分子的S—O键也从1.54 Å增大到1.73 Å,说明SO2分子在α-Fe2O3表面吸附后被激活,S原子与Fe原子间的键长为2.43 Å。
为深入解析SO2在α-Fe2O3(001)表面的吸附机制,计算了1A和1B的能带结构和局域态密度(PDOS)。图3表明,在吸附构型1A中的Fe-3d和O-2p轨道在能量区域内(-2.0 ~ 0.5 eV)、(-6.4~-3.2 eV)和(-11.8 ~-10.3 eV)有明显的重叠;在吸附构型1B中的Fe-3d和S-2p轨道在(-7.2~0.5 eV)和(-12.0 ~-10.6 eV)有明显的重叠。但是Fe-3d与S-2p轨道的重叠程度明显高于Fe-3d与O-2p轨道重叠程度,这表明SO2在α-Fe2O3(001)表面的吸附更倾向于通过d-p(Fe-3d-S-2p)轨道杂化作用,这一结果不仅与前面计算的吸附能结果一致,而且与Baltrusaitis等报道SO2在α-Fe2O3表面吸附的实验结果一致(Baltrusaitietal., 2007)。为进一步说明相互作用机制,计算了电荷密度和前线轨道说明吸附体系中电子的转移,结果如图3b和3c所示。由前线轨道可知SO2与α-Fe2O3(001)间的电子转移主要依赖于Fe原子和S原子之间,同时根据差分电荷密度和总电荷密度发现在Fe原子和S原子之间有明显的电子转移,由此说明Fe原子和S原子间的d-p(Fe-3d-S-2p)轨道杂化是SO2在α-Fe2O3(001)表面吸附的主要机制。
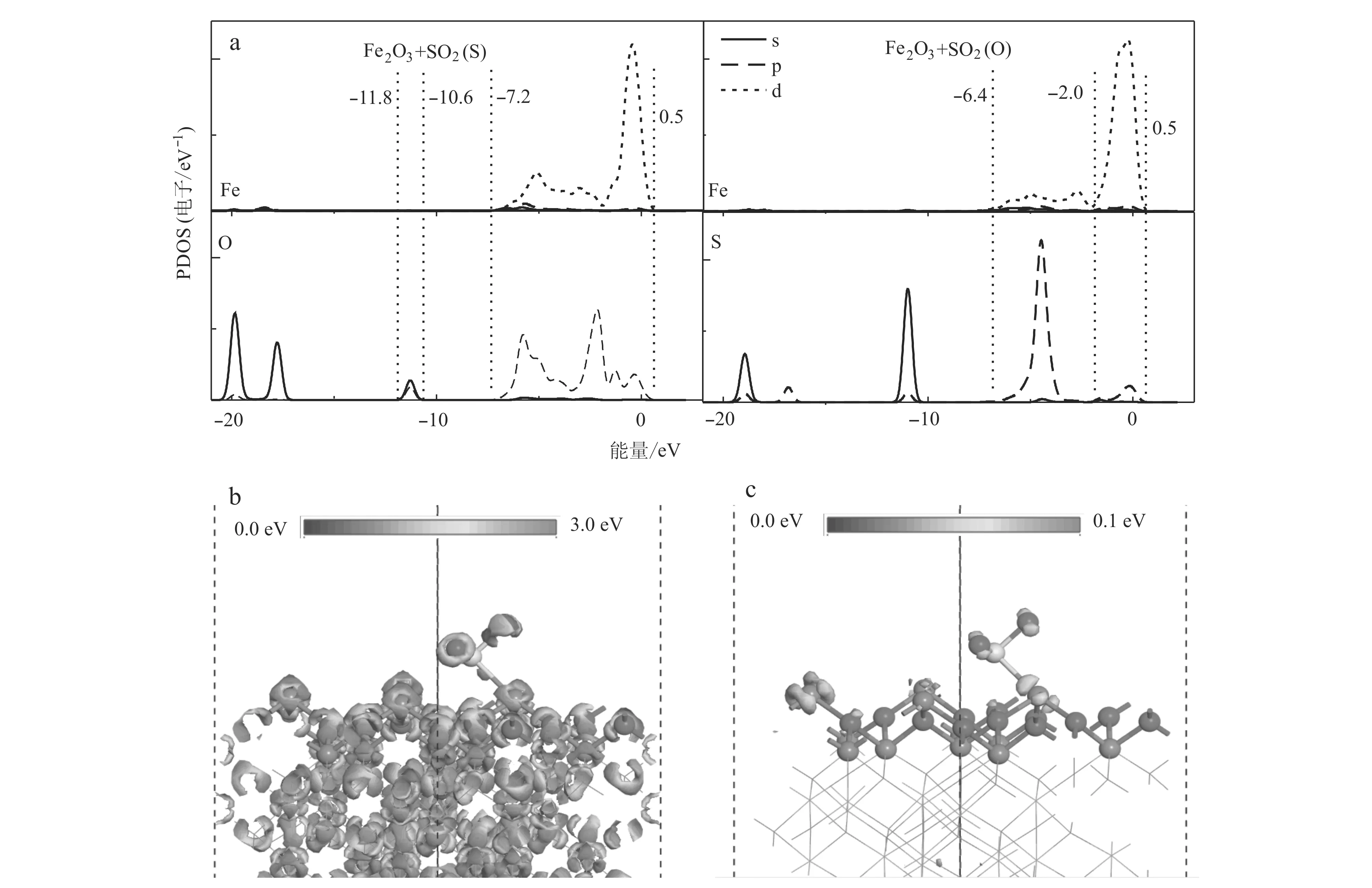
图 3 α-Fe2O3+ SO2不同吸附构型的PDOS图(a)、表面电荷密度图(b)和前线轨道图(c)Fig. 3 PDOS charts of different adsorption configurations of α-Fe2O3 + SO2 (a), surface charge density (b) and frontier orbital maps (c)
以Mulliken电荷分析作为定量分析电子转移的重要依据, 由表2中所示的Fe原子和S(或O)原子Mulliken电荷表明电子的转移是从α-Fe2O3表面向SO2转移, 吸附构型1A和1B的电子转移分别为0.61 e和0.43 e,说明吸附构型2A的相互作用强于吸附构型2B,这一结论与吸附能和能带结构分析结果一致。因此,α-Fe2O3(001)表面的Fe作为吸附SO2的活性位点,吸附构型2A作为SO2在α-Fe2O3(001)表面吸附的最优吸附方式,d-p(Fe-3d-S-2p)轨道杂化是Fe—S键形成的主要原因。
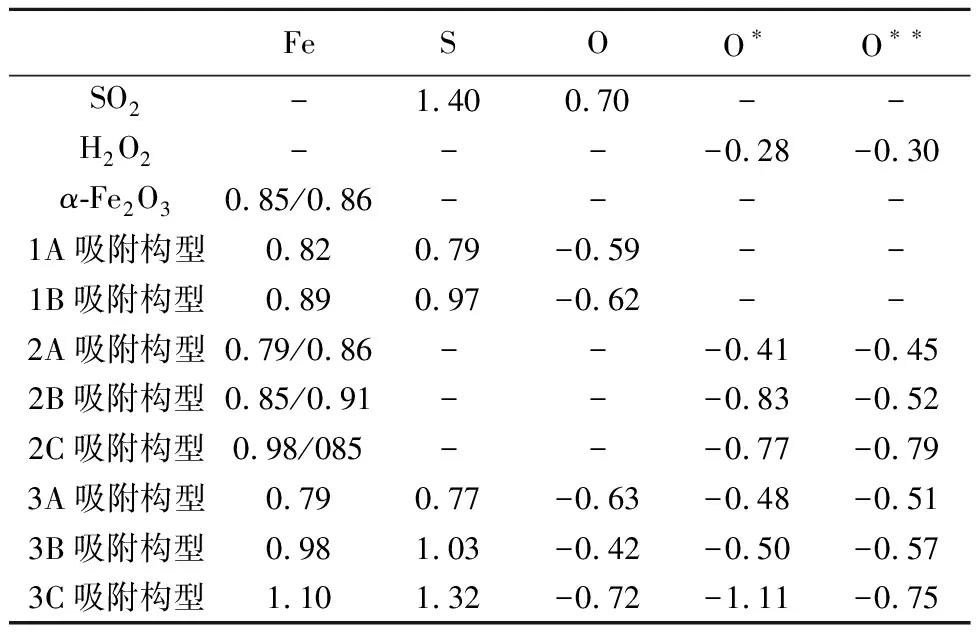
表 2 不同原子电荷Mulliken布局分析 eTable 2 Mulliken layout analysis of different atomic charges
注: O代表SO2中的O,O*和O**分别代表H2O2中的两个O。
2.2 H2O2在α-Fe2O3(001)表面的吸附与分解
H2O2能够在矿物氧化物上发生非均相分解产生·OH,具有氧化性的·OH基团氧化作用是氧化SO2的重要途径,H2O2在α-Fe2O3表面的分解可作为氧化性·OH基团产生的主要来源。因此,研究H2O2在α-Fe2O3(001)表面的吸附机制,可以为研究·OH基团氧化SO2提供依据。考虑到不同的吸附位点,得到了H2O2在α-Fe2O3(001)表面的吸附构型2A与分解吸附构型2B和2C。其中,吸附构型2A是H2O2分子通过O原子与表面的Fe原子吸附;分解吸附构型2B和2C是由H2O2分子分别分解为H2O+O和·OH+·OH与表面的Fe原子吸附,如图4所示。在吸附构型2A中,H2O2通过表面的铁原子分子平行吸附在α-Fe2O3(001)表面。H2O2与α-Fe2O3(001)表面不仅形成Fe—O键(1.95 Å),而且H2O2的两个氢原子与两个表面氧原子形成两个氢键的长度分别为2.89 Å和2.70 Å。与SO2的吸附相似,表面的Fe原子作为H2O2吸附的活性位点,H2O2的吸附是通过H2O2的O原子和表面的Fe原子。同时,在α-Fe2O3(001)表面吸附H2O2的O—O键相比于独立的H2O2明显增强(1.54 Å→2.85 Å)。为此,考虑了H2O2以不同的分解方式在α-Fe2O3(001)表面的吸附,得到了稳定的吸附构型2B和2C。吸附能计算表明吸附构型2C相比于吸附构型2A和2B具有较低的吸附能,由此说明H2O2在α-Fe2O3(001)表面的吸附方式是以两个·OH基团与和α-Fe2O3的Fe原子结合,并以此作为最佳的吸附构型。
为进一步描述H2O2与α-Fe2O3(001)表面间的吸附和分解机制,我们计算了吸附体系的能带结构和电荷密度。由图5a所示,能带结构(-20.0~0.8 eV)是由Fe-3d、O-2p轨道构成。对比发现在2A、2B和2C中H2O2的O-2p轨道发生了明显的变化,其中在吸附构型2C中O-2p轨道相比于吸附构型2A和2B发生了明显的轨道偏移和简并。O-2p轨道偏移和简并[-11.3~-1.6 eV (2A)和-8.4~-3.8 eV (2B)→-6.7~0.7 eV (2C)]增大了Fe-3d与·OH基团的O-2p轨道的重叠程度,增强了d-p(Fe-3d-O-2p)轨道杂化作用。同时,由差分电荷密度图和总电荷密度图可以看出羟基·OH基团的氧原子与羟基结合的两个铁原子间有明显的电荷转移积累。这一现象表明,电荷密度从Fe原子转移到了羟基的O原子,·OH与α-Fe2O3(001)表面发生了相互作用。由于在Fe2O3中只有2/3的八面体被Fe原子占据,这使得表面部分的Fe原子处于高电子组态。根据晶体场理论和能量最小原则,dz2和dx2+y2相比于dxy、dyz和dxz轨道指向配体,这使得Fe-3d与H2O2的O-2p轨道间的d-p(Fe-3dz2+O-2px和Fe-3dz2+O-2py)杂化方式转变为Fe-3d与·OH基团的O-2p轨道间的d-p(Fe-3dz2+O-2py)杂化方式。通过Mulliken电荷分析发现电子的转移是从α-Fe2O3表面向H2O2转移,吸附构型2A、2B和2C的电子转移分别为0.14 e、0.39 e和0.49 e。这说明吸附构型2C的相互作用强于吸附构型2A和2B。因此,Fe作为吸附H2O2的活性位点,吸附构型2C作为H2O2在α-Fe2O3(001)表面吸附的最优吸附方式,O-2p轨道的偏移和简并是增强d-p(Fe-3d-O-2p)轨道杂化的原因,也是吸附构型2C作为最优吸附方式的主要原因。
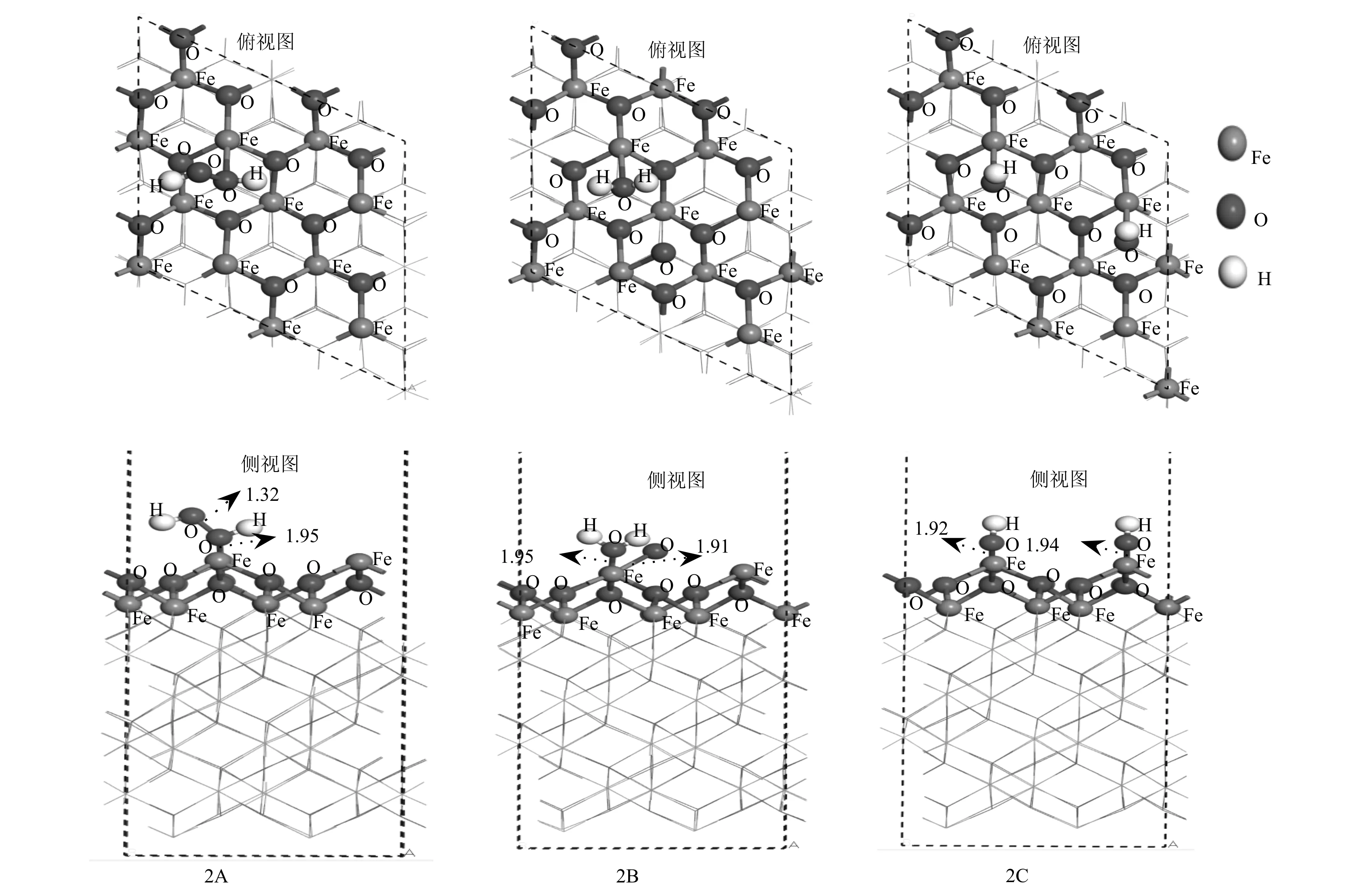
图 4 H2O2在α-Fe2O3(001)表面的吸附与分解构型Fig. 4 Adsorption and decomposition configuration of hydrogen dioxide on the surface of α-Fe2O3 (001)
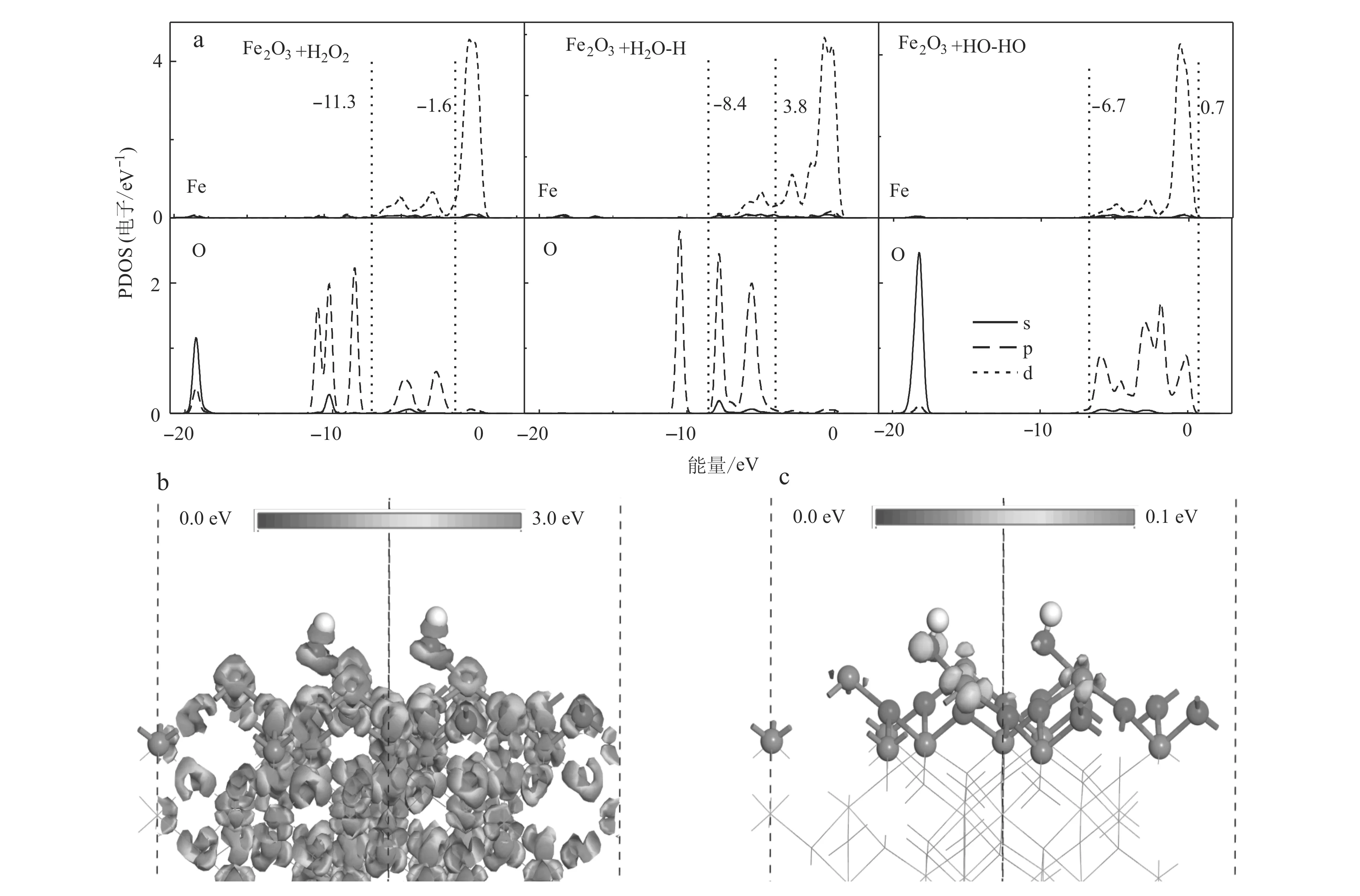
图 5 H2O2在α-Fe2O3 (001)表面吸附时不同吸附构型的PDOS图(a)、表面电荷密度(b)和前线轨道图(c)Fig. 5 PDOS diagrams of different adsorption configurations of hydrogen dioxide on the surface of α-Fe2O3 (001) (a), surface charge density (b) and frontier orbital maps (c)
2.3 SO2和H2O2在α-Fe2O3 (001)表面共吸附
已有研究表明,SO2在α-Fe2O3(001)表面可以被H2O2氧化(Huangetal., 2015)。然而,仅仅计算H2O2在α-Fe2O3(001)表面吸附时,不能直接证实·OH基团氧化性的存在,因此,又研究了SO2和H2O2在α-Fe2O3(001)表面的共吸附作用机制。考虑SO2和H2O2的竞争吸附和H2O2在表面的分解,分别得到了3A、3B和3C共吸附构型,如图6所示。在3A和3B中,SO2和H2O2分子被吸附到α-Fe2O3(001)表面的Fe原子。根据吸附构型3A(-3.38 eV)和3B(-2.29 eV),SO2和H2O2在α-Fe2O3(001)表面之间存在竞争性吸附,SO2分子相比于H2O2分子会优先吸附在α-Fe2O3(001)表面。在吸附构型3C中,H2O2在α-Fe2O3(001)表面分解形成两个表面·OH基团,其中一个·OH基团通过氢键与SO2反应形成·OH+SO2团簇分子,铁氧键Fe—O和氢键H—O的键长分别为1.91 Å和1.76 Å。同时,对比于SO2和H2O2共吸附时的吸附能,当H2O2分解为两个表面·OH基团与SO2在α-Fe2O3(001)表面具有较低的吸附能,说明SO2和H2O2在α-Fe2O3(001)表面是通过H2O2产生的·OH基团将SO2氧化,以·OH+SO2团簇分子的形式吸附在α-Fe2O3(001)表面。这与Huang等实验观察到H2O2与SO2在矿物粉尘上迅速反应,H2O2在矿物粉尘上的非均相分解产生的·OH自由基会立即与吸附的SO2反应相一致(Huangetal., 2015)。
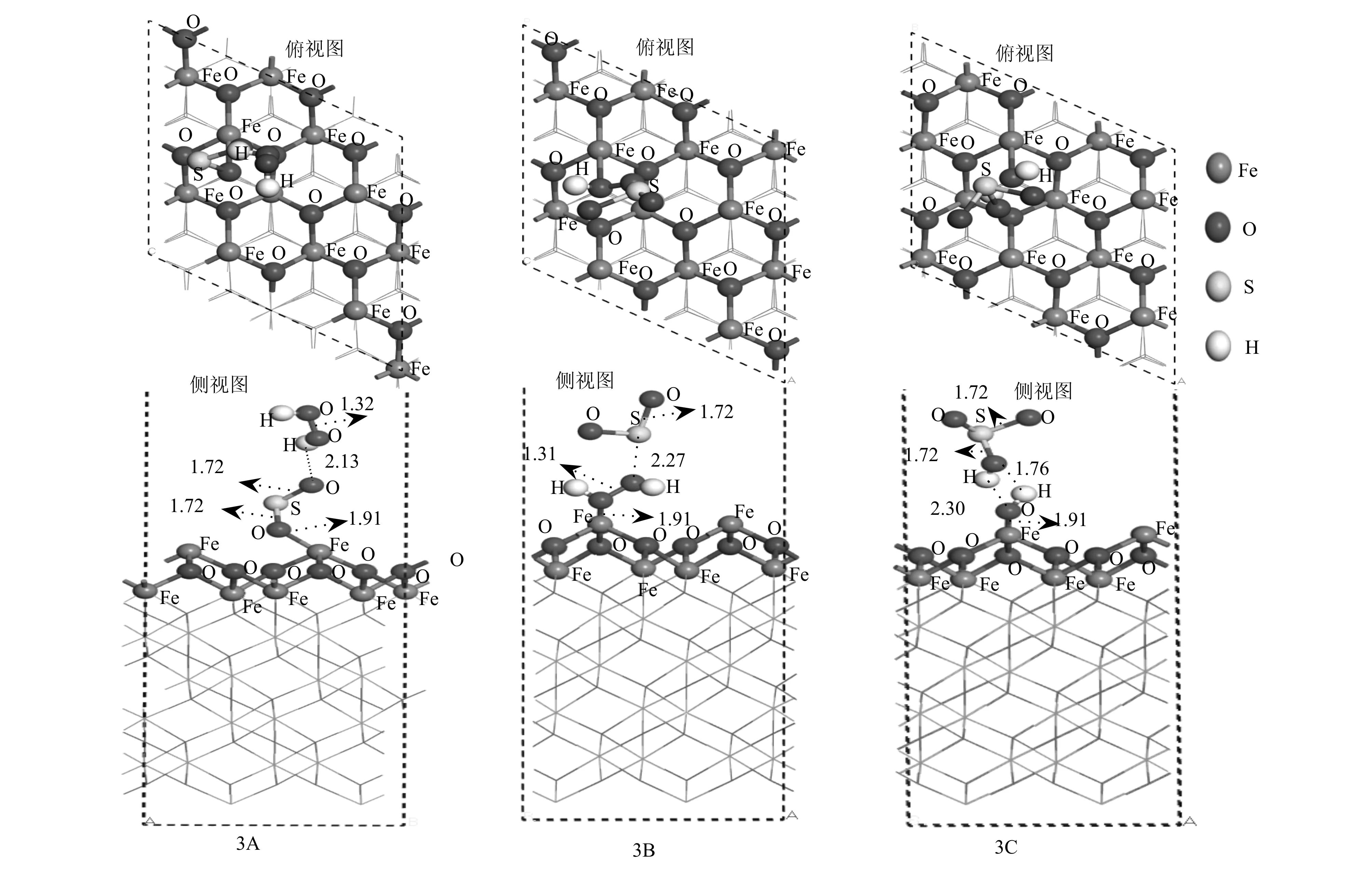
图 6 SO2和H2O2在α-Fe2O3(001)表面共吸附构型Fig. 6 Co-adsorption configuration of SO2 and H2O2 on α-Fe2O3 (001) surface
为进一步探讨SO2和H2O2在α-Fe2O3(001)表面的共吸附机制和·OH对SO2的氧化机制,我们计算了吸附构型3C的能带结构和电荷密度。如图7a所示,能带结构的价带(-20.0~0.8 eV)主要由Fe-3d、O-2p、S-2p轨道构成。对比发现在2·OH+α-Fe2O3(001)的O-2p4轨道H2O2+α-Fe2O3(001)发生了明显的劈裂,这增大了Fe-3d与·OH的O-2p轨道的重叠程度,改变了原来的d-p(Fe-3d-O-2p)轨道杂化强度。同时,在吸附构型3C中·OH基团的O-2p轨道与SO2的S-2p轨道在费米面(-7.5~0.8 eV)附近的重叠程度也明显增大,使得O-2p与S-2p轨道p-p(O-2p-S-2p)轨道杂化增强。差分电荷密度和总电荷密度计算表明,在构型3C中O和Fe原子(O—Fe键)与O和S原子(O—S键)周围有明显的电荷积聚,表明这些粒子之间有很强的相互作用。电子从表面Fe原子转移到·OH的O原子,形成一个O—Fe键;在·OH+SO2团簇分子中,由于高度局域化的O-2p轨道具有较高的电负性,诱导处于半满状态的S-2p轨道电子转移到另一个·OH的O原子的O-2p,形成一个S—O键,使得SO2被氧化[S(SO2)-电荷布局:0.79 e→1.32 e;O(H2O2)-电荷布局: -0.77 e→-1.11 e]形成·OH +SO2团簇。为进一步说明相互作用机制,进行了电荷的布局分析。通过Mulliken电荷分析发现电子的转移是从α-Fe2O3表面向H2O2分解产生的·OH转移,由SO2向H2O2分解得到的另一个·OH转移,其电荷转移(0.22 e)明显强于吸附构型3A和3B的电子转移(分别为0.07 e和0.10 e)。这表明吸附构型3C的相互作用强于吸附构型3A和3B。因此,吸附构型3C作为H2O2在α-Fe2O3(001)表面吸附的最优吸附方式。d-p(Fe-3d-O-2p)和p-p(S-2p-O-2p)轨道杂化方式的转变是H2O2分解的主要原因。
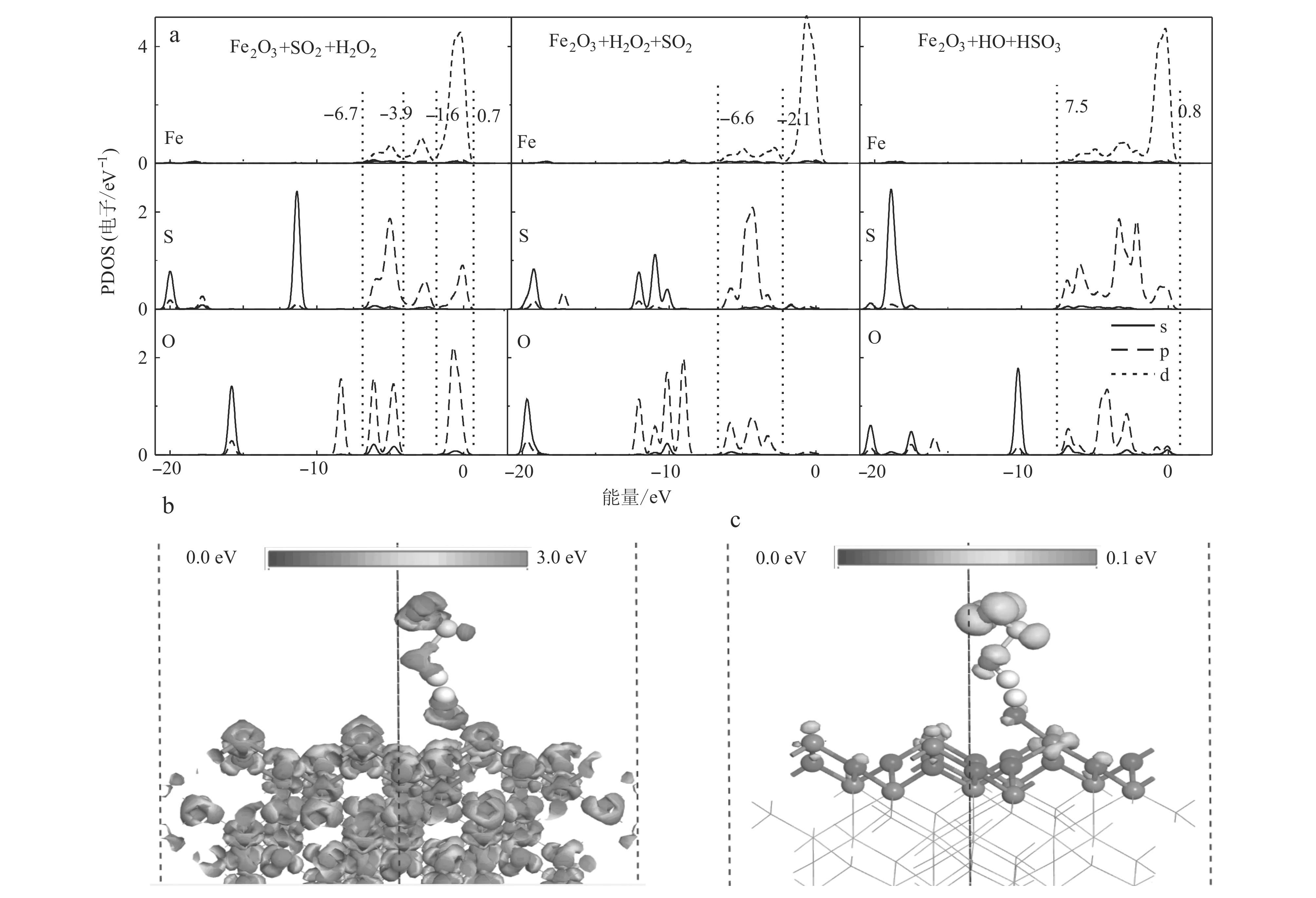
图 7 SO2和H2O2在α-Fe2O3(001)表面共吸附的不同吸附构型的PDOS图(a)、表面电荷密度图(b)和前线轨道图(c)Fig. 7 PDOS diagrams of different adsorption configurations for co-adsorption of SO2 and H2O2 on the surface of α-Fe2O3 (001) (a) , surface charge density (b) and frontier orbital (c) diagrams
3 结论
通过模拟计算SO2与H2O2在α-Fe2O3(001)表面的吸附机制,阐释了SO2在α-Fe2O3(001)表面氧化机制。计算结果表明,SO2、H2O2的吸附分别是通过SO2的S原子、H2O2的O原子与α-Fe2O3(001)表面的Fe原子间的d-p(SO2: Fe-3d-S-2p;H2O2: Fe-3d-O-2p)轨道发生杂化作用。特别地,H2O2在α-Fe2O3(001)表面的最优赋存形式是以两个·OH基团与α-Fe2O3(001)表面的Fe原子吸附。当SO2与H2O2在α-Fe2O3(001)表面共吸附时,H2O2分子以·OH基团优先吸附在α-Fe2O3(001)表面,一个·OH基团与吸附的SO2发生氧化反应,将SO2氧化为·OH+SO2团簇分子[S(SO2)-电荷布局: 0.79 e→1.32 e;O(H2O2)-电荷布局: -0.77 e→-1.11 e]。·OH+SO2团簇分子与另一个·OH基团通过氢键吸附在α-Fe2O3(001)表面。
