MiR-32-5p aggravates intestinal epithelial cell injury in pediatric enteritis induced by Helicobacter pylori
2019-11-21JingFengJianGuoJunPingWangBaoFengChai
Jing Feng, Jian Guo, Jun-Ping Wang, Bao-Feng Chai
Abstract BACKGROUND Pediatric enteritis is one of the infectious diseases in the digestive system that causes a variety of digestive problems, including diarrhea, vomiting, and bellyache in children. Clinically, Helicobacter pylori (H. pylori) infection is one of the common factors to cause pediatric enteritis. It has been demonstrated that aberrant expression of microRNAs (miRNAs) is found in gastrointestinal diseases caused by H. pylori, and we discovered a significant increase of miR-32-5p in H.pylori-related pediatric enteritis. However, the exact role of miR-32-5p in it is still unknown.AIM To investigate the role of aberrant miR-32-5p in pediatric enteritis induced by H.pylori.METHODS MiR-32-5p expression was detected by quantitative real time-polymerase chain reaction. The biological role of miR-32-5p in H. pylori-treated intestinal epithelial cells was evaluated by Cell Counting Kit-8 assay and flow cytometry. The potential target of miR-32-5p was predicted with TargetScanHuman and verified by luciferase assay. The downstream mechanism of miR-32-5p was explored by using molecular biology methods.RESULTS We found that miR-32-5p was overexpressed in serum of H. pylori-induced pediatric enteritis. Further investigation revealed that H. pylori infection promoted the death of intestinal epithelial cells, and increased miR-32-5p expression. Moreover, miR-32-5p mimic further facilitated apoptosis and inflammatory cytokine secretion of intestinal epithelial cells. Further exploration revealed that SMAD family member 6 (SMAD6) was the direct target of miR-32-5p, and SMAD6 overexpression partially rescued cell damage induced by H.pylori. The following experiments showed that miR-32-5p/SMAD6 participated in the apoptosis of intestinal epithelial cells induced by transforming growth factor-β-activated kinase 1 (TAK1)-p38 activation under H. pylori infection.CONCLUSION Our work uncovered the crucial role of aberrant expression of miR-32-5p in H.pylori-related pediatric enteritis, and suggested that the TAK1-p38 pathway is involved in it.
Key words: MiR-32-5p; SMAD family member 6; Transforming growth factor-βactivated kinase 1; Apoptosis; Enteritis; Helicobacter pylori
INTRODUCTION
Enteritis is a common disease of the digestive system in children among outpatients[1].Etiologically,Helicobacter pylori(H. pylori) infection is one of the most important pathogenic factors to induce pediatric enteritis[2]. The stomach is the primary organ that is damaged byH. pylori. However,H. pylori-induced enteritis is seen with increasing incidence in recent years.
H. pyloriinfection is regarded as a class I carcinogen[3]. Normally,H. pylori-induced gastritis could lead to gastric ulcer, which is the major precancerous lesion if without treatment. Although mainly residing in stomach,H. pyloridisplays a strong ability of acid resistance. As a pathogen,H. pyloricould attack and damage the mucosa of the digestive tract by recruiting and activating neutrophils[4], inducing abnormal expression of key proteins[5]and microRNAs (miRNAs)[6], and releasing cytotoxic substances[7]. A previous study showed thatH. pyloriinfection accounted for 6% of children with duodenitis[8]. Moreover, Gimigaet al[9]found that gastritis and duodenitis contributed to half of children with upper gastrointestinal bleeding, and 36.89% of participants were diagnosed withH. pyloriinfection. These findings suggested a relatively high prevalence of children withH. pyloriinfection in the digestive system.
MicroRNAs (miRNAs) belong to non-coding RNA molecules that are abundant in eukaryotic organisms[10,11]. MiRNAs have no ability to encode proteins, but contribute to the modulation of gene expression[11,12]. A recent study revealed the upregulation of miR-146a and miR-155 in patients with gastritis induced byH. pyloriinfection[13], with the similar findings demonstrated by another research group[14]. Cortés-Márquezet al[14]further grouped the patients with gastritis by age, and found that both children and adults withH. pylori-induced gastritis displayed higher levels of miR-145a and miR-155. Moreover, the cancerization tendency ofH. pylori-induced gastritis was correlated with the upregulation of miR-146a and miR-155[13,14]. In addition,H. pyloriinfection could result in the downregulation of miRNAs. The decreased expression of miR-24-3p was shown inH. pylori-induced gastritis tissue samples compared with the gastritis tissues withoutH. pyloriinfection[15]. Zouet al[16]demonstrated that gastric epithelial cells treated with miR-3178 mimic presented alleviated inflammation induced byH. pylori, and miR-3178 could blockH. pylori-induced carcinogenesis by targeting the TRAF1-NF-κB pathway. Therefore, the relationship between miRNAs andH. pyloriinfection is not positively correlated. Except for causing gastric epithelial cell damageviaabnormal expression of miRNAs,H. pyloriinfection-induced miRNAs might contribute to intestinal epithelial cell damage. However, little is currently known about miRNAs andH. pylori-related enteritis.
Herein, our research revealed significant upregulation of miR-32-5p by targeting SMAD family member 6 (SMAD6) inH. pylori-infected intestinal epithelial cells,which aggravated the damage ofH. pylorito the cells and might contribute to the pathogenesis of pediatric enteritis induced byH. pylori.
MATERIALS AND METHODS
Participants
Serum samples were obtained from children withH. pylori-induced enteritis (n= 15)and healthy controls (n= 15), and the participants were from Shanxi Provincial People’s Hospital. Procedures in this study were approved by the Ethics Committee of Shanxi University and Ethics Committee of Shanxi Provincial People’s Hospital,and complied with the guidelines of Declaration of Helsinki. Both guardians of the children and the participants were informed of the purpose of the study, and signed an informed consent form.
Cell culture and H. pylori strain
Human intestinal epithelial cell line HIEC-6 was cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS). Human embryonic kidney cell line HEK-293T was cultured in DMEM medium with 10% FBS. The medium and FBS were purchased from ThermoFisher Scientific (United States).H. pyloristrain T81213-NTB (ATCC 46396) was cultured as previously described[17]. The concentration ofH. pyloriwas adjusted to 1 × 109CFU/mL, and 1 × 106CFU/mL was used in our experiments.
Quantitative real time-polymerase chain reaction
Serum miRNAs were extracted using a miRNeasy Serum/Plasma Kit (QIAGEN,Germany), miRNAs of cells were obtained with a miRNeasy Mini Kit (QIAGEN,Germany), and total RNA of cells was extracted using TRIzol (Takara, Japan). cDNA was acquired from the extracted RNA, and quantitative real time-polymerase chain reaction (qRT-PCR) was carried out by using a SYBR Premix Ex Taq II Kit (Takara,Japan). The expression level was calculated by using 2-ΔΔCTmethods. Primers used in our study are displayed in Table 1.
Cell transfection
SiRNAs, miRNA mimic and inhibitor, and overexpression vectors were obtained from Sangon Biotech (China). Cell transfection was carried out with Lipofectamine 3000(Invitrogen, United States) according to the manufacturer’s instructions. The sequences used are shown in Table 1.
Detection of cell viability
Cell Counting Kit-8 (Beyotime, China) assay was utilized to evaluate cell viability according to the manufacturer’s instructions. Cells were seeded followed by cell transfection in the presence or absence ofH. pylori. Ten microliters of the reagents were added into each well at 37 °C for 1.5 h. The value of optical density (OD) was detected with a microplate reader (Bio-Rad, United States) at 450 nm.
Flow cytometry
After cell transfection, cells were incubated with TAK1 inhibitor NG25 (5 μmol/L,APExBIO, United States) and p38 inhibitor (10 μmol/L, Cell Signaling Technoloby,United States) in the presence or absence ofH. pylori, harvested, and stained using an Annexin V-FITC/PI apoptosis kit (Beyotime, China) according to the manufacturer’s instructions. The apoptotic rate is represented as the positive rate of cells labelled with Annexin V, and the results were analyzed by flow cytometry (Accuri C6, United States).
Luciferase assay
HEK-293T cells were used to validate the binding between miR-32-5p and SMAD6.Wild type (WT) and mutant (Mut) SMAD6 containing predicted sites with miR-32-5p were obtained from Sangon Biotech (China). After transfection for 48 h, the cells were tested using the Bio-Glo Luciferase Assay System (Promega, United States).

Table 1 Sequences of primers, siRNAs, microRNA mimic, and microRNA inhibitor used in this study
Western blot analysis
Western blot analysis was performed as previously described[16]. Primary antibodies(mouse monoclonal to SMAD6, Santa Cruz Biotechnology, United States; rabbit monoclonal to TAK1, Abcam, United States; rabbit monoclonal to TAK1, phosphor S439, Abcam; rabbit monoclonal to p38, Abcam; rabbit monoclonal to p38, phosphor T180, Abcam; mouse monoclonal to Actin, Beyotime, China) were used in our research. The ChemDoc XRS+ System (Bio-Rad, United States) was utilized to evaluate the protein bands.
Statistical analysis
The data are expressed as the mean ± standard deviation (SD) and analyzed by nonparametrict-tests using GraphPad Prism 6.0 software (GraphPad, United States).P<0.05 was considered statistically significant.
RESULTS
MiR-32-5p is overexpressed in enteritis
To explore the potential role of miR-32-5p in pediatric enteritis, we first monitored the expression of miR-32-5p. After separating the serum from children with enteritis induced byH. pyloriand healthy controls, we found that miR-32-5p was upregulated in serum of children withH. pylori-induced pediatric enteritis (Figure 1A). Next, we treated intestinal epithelial cells withH. pylori. The qRT-PCR results displayed thatH.pyloriled to a significant increase of miR-32-5p in intestinal epithelial cells (Figure 1B).These findings suggested that miR-32-5p might play a crucial role inH. pylori-related pediatric enteritis.
MiR-32-5p regulates biological behavior of H. pylori-treated intestinal epithelial cells
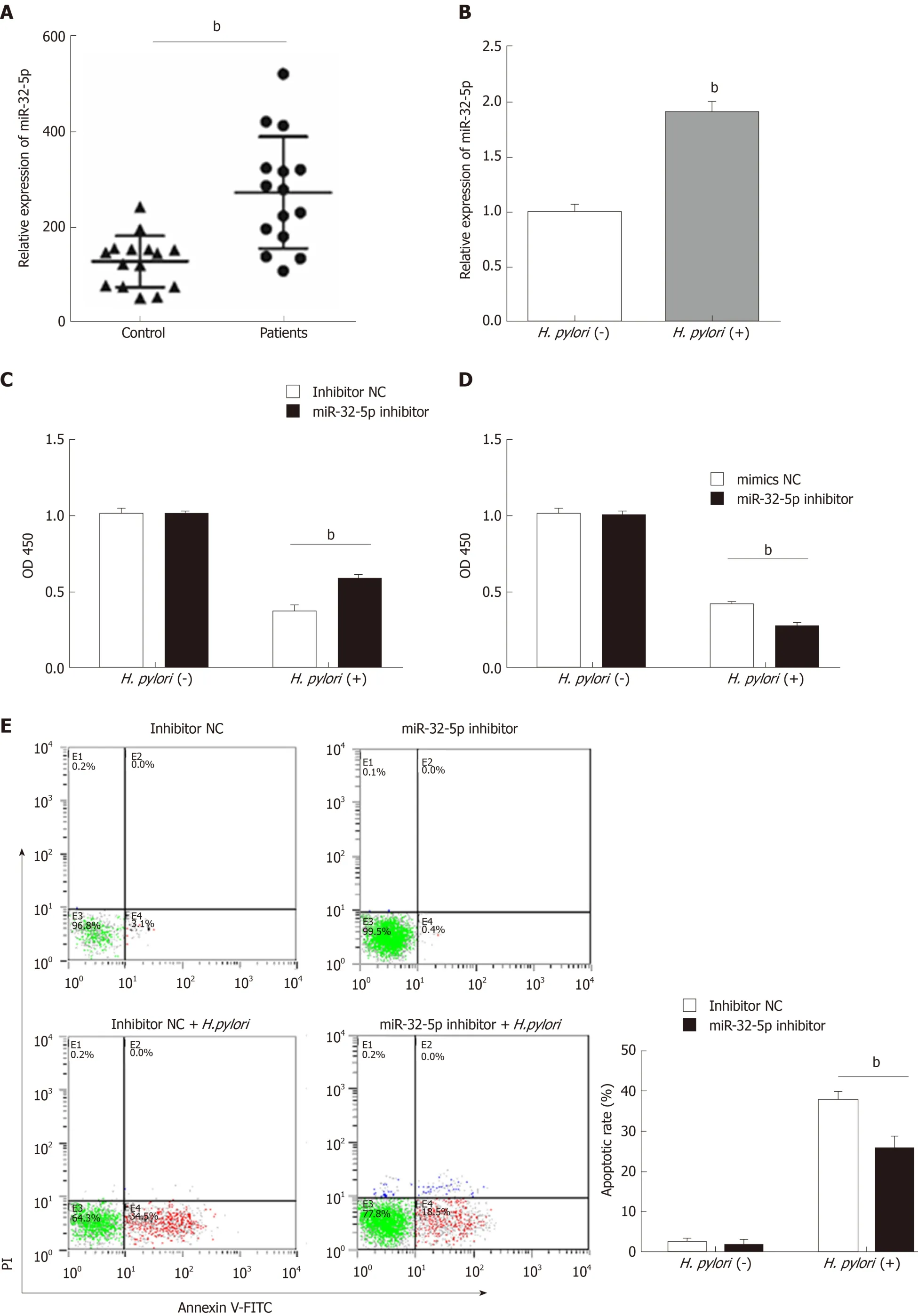
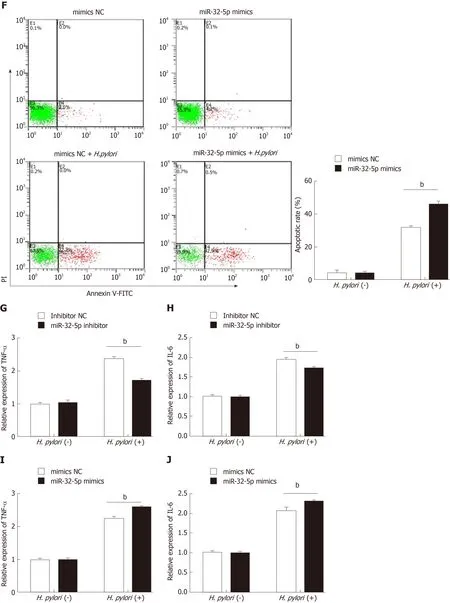
Figure 1 Aberrant expression of miR-32-5p in enteritis regulates biological function of intestinal epithelial cells. A: Expression of miR-32-5p in serum of pediatric enteritis; B: Expression of miR-32-5p in intestinal epithelial cells infected by Helicobacter pylori (H. pylori); C: Cell viability measurement in intestinal epithelial cells transfected with miR-32-5p inhibitor in the presence or absence of H. pylori; D: Viability measurement in intestinal epithelial cells transfected with miR-32-5p mimic in the presence or absence of H. pylori; E: Apoptosis of intestinal epithelial cells with miR-32-5p inhibitor transfection in the presence or absence of H. pylori; F:Apoptosis of intestinal epithelial cells with miR-32-5p mimic transfection in the presence or absence of H. pylori; G: The mRNA level of TNF-α in H. pylori-infected intestinal epithelial cells in the presence of miR-32-5p inhibitor; I: The mRNA level of TNF-α in H. pylori-infected intestinal epithelial cells in the presence of miR-32-5p mimic; H: The mRNA level of IL-6 in H. pylori-infected intestinal epithelial cells in the presence of miR-32-5p inhibitor; J: The mRNA level of IL-6 in H. pylori-infected intestinal epithelial cells in the presence of miR-32-5p mimic. bP < 0.01. H. pylori: Helicobacter pylori.
Next, cell transfection with miR-32-5p inhibitor or miR-32-5p mimic was utilized to verify the role of miR-32-5p in cell viability in the presence or absence ofH. pylori. We found thatH. pyloriimpaired cell viability, and miR-32-5p inhibitor partially restored the viability ofH. pylori-infected intestinal epithelial cells (Figure 1C). On the contrary,miR-32-5p mimic further caused the decrease of viability ofH. pylori-infected intestinal epithelial cells (Figure 1D). In parallel,H. pylori-infected intestinal epithelial cells with miR-32-5p inhibitor displayed a lower apoptotic rate, and miR-32-5p mimic facilitated apoptosis ofH. pylori-infected intestinal epithelial cells (Figure 1E and F). In addition, we found that TNF-α was upregulated in intestinal epithelial cells infected withH. pylori, which was decreased by miR-32-5p inhibitor transfection and further increased by miR-32-5p mimic (Figure 1G and I). IL-6 expression displayed the similar trend as TNF-α (Figure 1H and J).
SMAD6 is the direct target of miR-32-5p
To further explore the downstream mechanism of miR-32-5p, we used TargetScanHuman 7.2 (http://www.targetscan.org/vert_72/) to identify SMAD6,acting as a mediator of anti-inflammatory activity, as the potential target of miR-32-5p. The luciferase assay showed that SMAD6 was the direct target of miR-32-5p(Figure 2A). After transfecting with miR-32-5p mimic into intestinal epithelial cells,we found that the protein level of SMAD6 was downregulated (Figure 2B). Next, we performed cell transfection assay to make SMAD6 overexpress and knock down(Figure 2C). We further found that SMAD6 overexpression ameliorated the death of intestinal epithelial cells infected byH. pylori(Figure 2D). On the contrary, SMAD6 siRNA acceleratedH. pylori-induced cell death (Figure 2E). In addition, SMAD6 could partially restrain the elevation of both TNF-α and IL-6 in intestinal epithelial cells infected byH. pylori(Figure 2F and G), while SMAD6 knockdown exerted an opposite role as SMAD6 overexpression did in the expression of TNF-α and IL-6 (Figure 2H and I). Therefore, these findings suggested that SMAD6 played a critical role inH.pylori-infected intestinal epithelial cells by acting as the downstream molecule of miR-32-5p.
TGF-β1/p38 pathway is involved in apoptosis of H. pylori-infected intestinal epithelial cells
SMAD6 is a downstream molecule of transforming growth factor-β1 (TGF-β1), and transforming growth factor-β-activated kinase 1 (TAK1)-p38 cascade activation could be executed by TGF-β1[18,19]. We found that TGF-β1 treatment increased the apoptosis of intestinal epithelial cells, and both TAK1 inhibitor and p38 inhibitor could limit TGF-β1-induced apoptosis (Figure 3A). What’s more, TGF-β1 led to the upregulation of phosphorylated TAK1 (p-TAK1) and phosphorylated p38 (p-p38), which could be restrained by TAK1 inhibitor (Figure 3B). However, TGF-β1-mediated phosphorylation of TAK1 was not inhibited by p38 inhibitor (Figure 3B). As shown in Figure 3C and D, TAK1 inhibitor and p38 inhibitor could partially increase theviability of intestinal epithelial cells infected withH. pylori, while miR-32-5p mimic, combined with either TAK1 inhibitor or p38 inhibitor, led to a marked decrease in the cell viability (Figure 3C). On the contrary, miR-32-5p further increased cell viability with either TAK1 inhibitor or p38 inhibitor underH. pyloriinfection(Figure 3D). Consistent with the findings in the cell viability, we found that both TAK1 inhibitor and p38 inhibitor could partially restrained the apoptosis ofH.pylori-infected intestinal epithelial cells (Figure 3E and F). When miR-32-5p mimic was transfected into the cells with either TAK1 inhibitor or p38 inhibitor underH.pyloriinfection, apoptosis increased significantly (Figure 3E). However, miR-32-5p inhibitor transfection played an opposite role as miR-32-5p mimic did (Figure 3F).Thus, these results suggested that TGF-β1-TAK1-p38 cascade contributed to intestinal epithelial cell damage underH. pyloriinfection.
MiR-32-5p/SMAD6 contributes to TAK1-p38 pathway activation in intestinal epithelial cells infected by H. pylori
Next, we found the upregulation of p-TAK1 and p-p38 in intestinal epithelial cells withH. pyloriinfection (Figure 4A). SMAD6 is one of the inhibitory SMADs that could block TGF-β1 signaling[19]. Once SMAD6 overexpression plasmid was transfected into the cells, the activation of TAK1-p38 cascade was limited significantly (Figure 4B).Moreover, we stimulated intestinal epithelial cells with patient serum, and found that SMAD6 was downregulated (Figure 4C). When miR-32-5p antagonist was added into the patient serum, the downregulated expression of SMAD6 was reversed (Figure 4D). We also found that patient serum treatment activated TAK1 and p38 in intestinal epithelial cells compared with healthy control serum treatment (Figure 4E). However,miR-32-5p antagonist could result in downregulation of p-TAK1 and p-p38 in intestinal epithelial cells cultured in medium containing patient serum (Figure 4F).
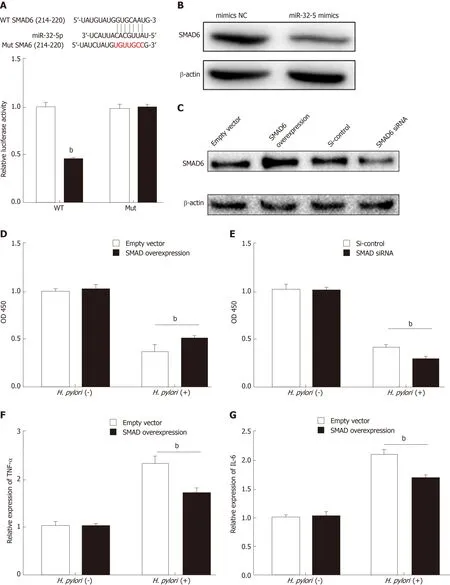
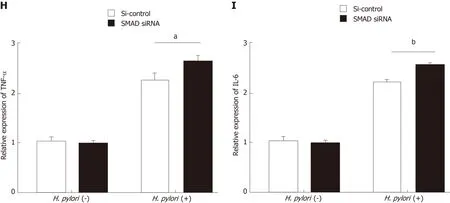
Figure 2 SMAD family member 6 is sponged by miR-32-5p in intestinal epithelial cells. A: Luciferase assay for determining the binding between miR-32-5p and SMAD family member 6 (SMAD6); B: Protein level of SMAD6 in intestinal epithelial cells transfected with miR-32-5p mimic; C: Expression of SMAD6 in intestinal epithelial cells after SMAD6 overexpression and knockdown; D: Cell viability measurement in intestinal epithelial cells after SMAD6 overexpression in the presence or absence of H. pylori; E: Cell viability measurement in intestinal epithelial cells after SMAD6 knockdown in the presence or absence of Helicobacter pylori (H. pylori); F:The mRNA level of TNF-α in H. pylori-infected intestinal epithelial cells after SMAD6 overexpression; H: The mRNA level of TNF-α in H. pylori-infected intestinal epithelial cells after SMAD6 knockdown; G: IL-6 expression in H. pylori-infected intestinal epithelial cells after SMAD6 overexpression; I: IL-6 expression in H. pyloriinfected intestinal epithelial cells after SMAD6 knockdown. aP < 0.05, bP < 0.01. SMAD6: SMAD family member 6; H. pylori: Helicobacter pylori.
DISCCUSION
As a common digestive disease in children, enteritis could cause abdominal pain,diarrhea, and dyspepsia. If chronic enteritis results from the nonstandard treatment,children might display malnutrition, even severely affecting health and growth of children. Virus infection is regarded as the leading cause of pediatric enteritis worldwide[20,21], which might lead to a local outbreak. However, what could not be ignored is that bacterial infection is also an important pathogenic factor resulting in pediatric enteritis. It has been reported thatH. pyloriinfection displayed a remarkable correlation with pediatric enteritis[2,22]. However, there is a lack of systematic understanding about the correlation betweenH. pyloriinfection and the pathogenesis of pediatric enteritis.
H. pyloriis a common bacterium that could cause gastritis, gastric ulcer, and even gastric cancer[3,7,23]. A great deal of evidence has revealed that multiple factors contribute toH. pyloriinfection, including autophagy, miRNAs, and long non-coding RNAs (lncRNAs).H. pyloricould promote autophagy of gastric adenocarcinoma epithelial cells, and increased antibiotic resistance ofH. pylori-infected cells[24]. Yanget al[25]reported that miR-30d regulated autophagy-related proteins to facilitateH. pylorisurvival within cells. An increasing number of miRNAs and lncRNAs were identified by different research groups, which displayed a tight relationship withH. pyloriinfection[6,14,26,27]. In the current study, we found the upregulation of miR-32-5p in serum of patients withH. pylori-related pediatric enteritis andH. pylori-infected intestinal epithelial cells. In addition, miR-32-5p inhibition could rescue intestinal epithelial cells infected withH. pylori, reduceH. pylori-induced apoptosis, and restrain the increase of inflammatory cytokines ofH. pylori-infected cells, which inferred that miR-32-5p contributed to intestinal epithelial cell injury caused byH. pyloriinfection.
Currently, only a few studies focus on miR-32-5p, and these research findings are largely about the relationship between miR-32-5p and the pathogenesis of tumors,including cervical cancer[28], prostate cancer[29], hepatocellular carcinoma[30], and pancreatic cancer[31]. Besides, Zhang and colleagues[32]discovered thatMycobacterium tuberculosisinfection led to a significant increase of miR-32-5p in macrophages, which in turns promotedMycobacterium tuberculosissurvival in the infected cells by targeting the downstream molecules of miR-32-5p. We found that SMAD6 was a direct target of miR-32-5p to be involved inH. pylori-induced intestinal epithelial cell damage. It has been demonstrated that SMAD6 is one of the two inhibitory SMADs (e.g., SMAD6 and SMAD7) downstream of TGF-β, and could inhibit inflammation and cell apoptosis[19,33,34]. Our findings revealed that SMAD6 overexpression could increase resistance of intestinal epithelial cells toH. pylori, and reduce inflammatory cytokines caused byH. pylori. Next, we found that TGF-β1-activated TAK1-p38 cascade contributed toH. pylori-induced intestinal epithelial cell injury. Luo and colleagues reported that TGF-β1 mediatedH. pylori-related gastritis, and blocking TGF-β1 could effectively alleviate gastritis[35]. As we verified, inhibition of TAK1 activation and p38 activation could partially protect epithelial cells fromH. pyloriinfection. Moreover,miR-32-5p inhibition further improved the protective role of TAK1 inhibitor and p38 inhibitor inH. pylori-infected intestinal epithelial cells.
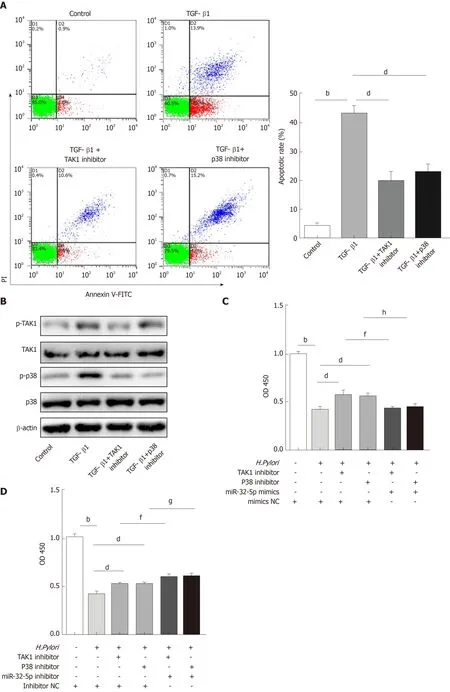
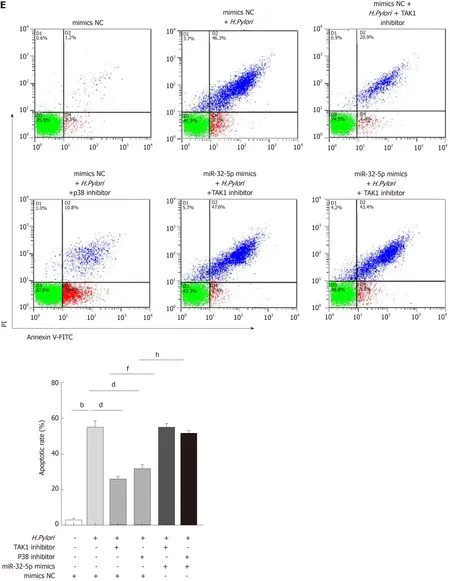
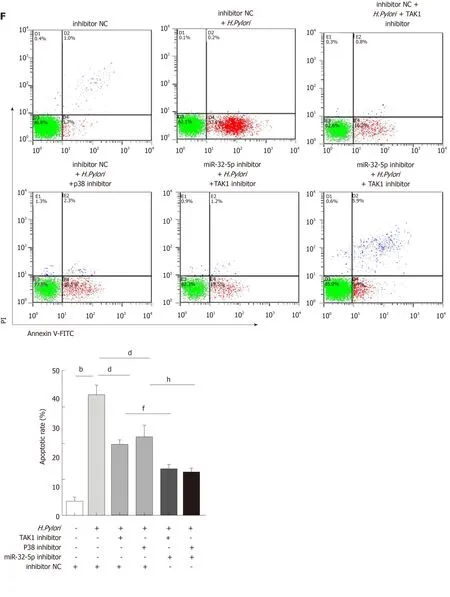
Figure 3 Transforming growth factor-β1/p38 participates in apoptosis of intestinal epithelial cells infected by Helicobacter pylori. A: Apoptosis detection in transforming growth factor-β1 (TGF-β1)-treated intestinal epithelial cells in the presence of transforming growth factor-β-activated kinase 1 (TAK1) inhibitor and p38 inhibitor; B: Protein levels of total TAK1, p38, phosphorylated TAK1, and phosphorylated p38 in TGF-β1-treated intestinal epithelial cells in the presence of TAK1 inhibitor and p38 inhibitor; C: Cell viability measurement in Helicobacter pylori (H. pylori)-infected intestinal epithelial cells with TAK1 inhibitor and p38 inhibitor treatment followed by transfection with miR-32-5p mimic; D: Cell viability measurement in H. pylori-infected intestinal epithelial cells with TAK1 inhibitor and p38 inhibitor treatment followed by transfection with miR-32-5p inhibitor; E: Apoptosis evaluation in H. pylori-infected intestinal epithelial cells with TAK1 inhibitor and p38 inhibitor treatment followed by transfection with miR-32-5p mimic; F: Apoptosis evaluation in H. pylori-infected intestinal epithelial cells with TAK1 inhibitor and p38 inhibitor treatment followed by transfection with miR-32-5p inhibitor. bP < 0.01, dP < 0.01, fP < 0.01, gP < 0.05, hP < 0.01. TGF-β1: Transforming growth factor-β1;TAK1: Transforming growth factor-β-activated kinase 1; H. pylori: Helicobacter pylori.
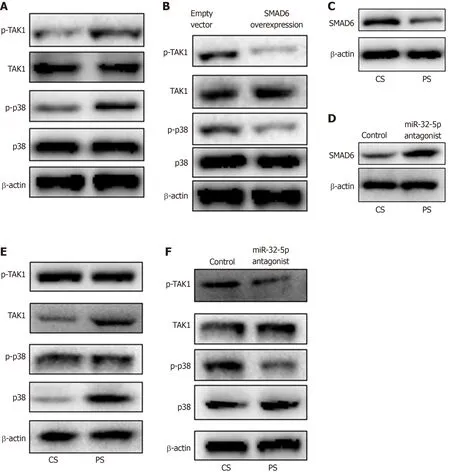
Figure 4 MiR-32-5p/SMAD family member 6 is involved in transforming growth factor-β-activated kinase 1-p38 pathway in intestinal epithelial cells. A:Detection of transforming growth factor-β-activated kinase 1 (TAK1)-p38 activation in Helicobacter pylori (H. pylori)-treated intestinal epithelial cells by Western blot; B:Inhibition of TAK1-p38 activation in H. pylori-treated intestinal epithelial cells with SMAD6 overexpression as revealed by Western blot; C: SMAD6 expression in intestinal epithelial cells treated with control serum and patient serum (PS); D: SMAD6 expression in PS-treated intestinal epithelial cells with miR-32-5p antagonist transfection; E: TAK1-p38 activation in PS-treated intestinal epithelial cells as revealed by Western blot; F: Inhibition of TAK1-p38 activation in PS-treated intestinal epithelial cells with miR-32-5p antagonist transfection as revealed by Western blot. CS: Control serum; PS: Patient serum; SMAD6: SMAD family member 6; TAK1:Transforming growth factor-β-activated kinase 1; H. pylori: Helicobacter pylori.
Recently, TGF-β was determined to be involved inH. pyloriinfection. A recent study found thatH. pylorifacilitated the expression of TGF-β, and induced downregulation of cystic fibrosis transmembrane conductance regulator (CFTR) and solute linked carrier 26 gene family A6 (SLC26A6) in the duodenum[36]. Patients withH. pylori-induced duodenal ulcers displayed significantly decreased expression of CFTR and SLC26A6, which displayed a remarkable correlation with the severity of the illness[37]. In addition to affecting the function of intestinal epithelial cells,H. pyloricould modulate the role of immune cells in the intestinal tract. A previous study suggested thatH. pyloriled to the upregulation of heat shock protein 60 in macrophages to promote TGF-β production, which contributed to the infiltration of regulatory T cells (Treg) to cause persistent infection[38]. Clinical data showed an increased number of Treg in children withH. pyloriinfection, and TGF-β had a positive correlation with the increase of Treg[39]. In our study, we discovered a novel manner involving TGF-β1 in whichH. pyloriinfection damaged intestinal epithelial cells. Induction of miR-32-5p byH. pyloriinfection led to decreased expression of SMAD6, which weakened the inhibitory role of SMAD6 in TGF-β1-TAK1-p38 cascade activation, as demonstrated by the treatment of serum samples in intestinal epithelial cells.
Based on the above findings, we concluded thatH. pyloriinfection promotes miR-32-5p upregulation to aggravate apoptosis and inflammation in intestinal epithelial cells by targeting SMAD6 and activating the TAK1-p38 pathway, highlighting a crucial role of miR-32-5p in the pathogenesis of pediatric enteritis caused byH. pylori.
ARTICLE HIGHLIGHTS
Research background
Helicobacter pylori(H. pylori) infection is a global issue that could cause a variety of diseases involving multiple organs. It is worth noting that the incidence ofH. pylori-related enteritis in children increases, but the underlying mechanism is largely unknown. It has been reported that miR-32-5p is overexpressed in diseases associated with bacterial infection. However, the potential role of miR-32-5p inH. pylori-induced pediatric enteritis is not clear.
Research motivation
To investigate the exact role of miR-32-5p in the pathogenesis of pediatric enteritis withH. pyloriinfection, and to find a novel target forH. pylori-related enteritis in children.
Research objectives
To explore the aberrant expression and significance of miR-32-5p in children withH. pylorirelated enteritis, especially in the damage of intestinal epithelial cells withH. pyloriinfection.
Research methods
qRT-PCR was performed to detect the expression of miR-32-5p in clinical samples andH. pyloriinfected intestinal epithelial cells. Cell Counting Kit-8 assay and flow cytometry were conducted to evaluate the role of miR-32-5p inH. pylori-infected intestinal epithelial cells.TargetScanHuman database and luciferase assay were utilized to verify the potential target of miR-32-5p. Western blot was employed to clarify the underlying mechanism of miR-32-5p in influencingH. pylori-infected intestinal epithelial cells.
Research results
The present study discovered the aberrant expression of miR-32-5p in pediatric enteritis withH.pyloriinfection andH. pylori-treated intestinal epithelial cells. Thein vitroexperiments showed the significance of miR-32-5p in regulating cell viability and apoptosis ofH. pylori-treated intestinal epithelial cells. We identified that SMAD family member 6 (SMAD6) was the direct target of miR-32-5p, and SMAD6 partially counteracted the harmful role of miR-32-5p by inhibiting the activation of TAK1-p38 cascade. However,in vivoassays are needed to further verify ourin vitrofindings and significance of miR-32-5p inH. pylori-induced pediatric enteritis.
Research conclusions
Our research first identified the upregulation of miR-32-5p inH. pylori-related pediatric enteritis.Further exploration revealed that miR-32-5p inhibited SMAD6 to activate the TAK1-p38 signaling pathway, aggravatingH. pylori-induced damage of intestinal epithelial cells. MiR-32-5p might be a potential target to overcomeH. pylori-induced damage of intestinal epithelial cells in children.
Research perspectives
Based on the clinical findings andin vitroexperiments, miR-32-5p could be a novel therapeutic target forH. pylori-induced damage of intestinal epithelial cells in children. Furtherin vivoassays are of necessity to clarify the deleterious effects of miR-32-5p onH. pylori-infected intestinal epithelial cells, which is very meaningful and would contribute to clinical application.
杂志排行

World Journal of Gastroenterology的其它文章
- lncreased circulating circular RNA_103516 is a novel biomarker for inflammatory bowel disease in adult patients
- Blood parameters score predicts long-term outcomes in stage II-III gastric cancer patients
- Tumor-infiltrating platelets predict postoperative recurrence and survival in resectable pancreatic neuroendocrine tumor
- Bacterobilia in pancreatic surgery-conclusions for perioperative antibiotic prophylaxis
- Yinchenhao decoction attenuates obstructive jaundice-induced liver injury and hepatocyte apoptosis by suppressing protein kinase RNA-like endoplasmic reticulum kinase-induced pathway
- Therapeutic potential of menstrual blood stem cells in treating acute liver failure