吡咯烷酮类螺环化合物与β-分泌酶活性中心的结合机制
2019-11-19刘中婷钟谢斌王汐璆庄文昌朱文友
刘中婷,钟谢斌,王汐璆,庄文昌,朱文友
(徐州工程学院 化学化工学院,江苏 徐州 221111)
阿尔茨海默症(AD)俗称老年痴呆,进行性记忆丧失和认知能力下降是该病的典型临床特征,其主要病理特征是存在神经纤维缠结和细胞外淀粉样蛋白“斑块”[1]。斑块主要由淀粉样蛋白前体蛋白(APP)的蛋白水解片段组成。随着对阿尔茨海默症的不断深入研究,人们发现β-分泌酶(BACE)是阿尔茨海默病患者脑中斑块主要成分淀粉样蛋白前体蛋白水解片段形成的一种关键酶。AD 的发生和发展与大脑中斑块的沉积有直接关系,抑制BACE1的水解作用将会阻断或减慢斑块的产生和聚集,从而减缓AD的发生和发展,起到干预治疗该疾病的作用[2-4]。抑制BACE1的水解作用已成为阿尔茨海默病患者药物干预的主要手段[5]。
早期的BACE抑制剂都是肽类药物,其作用主要是截取APP断裂位点附近的片段,通过加入非天然的氨基酸,来模拟BACE1与APP作用的过渡态异构体,从而与APP发生竞争性抑制,减少β淀粉样多肽(Aβ)的产生[6]。虽然肽类抑制剂具有良好的生物相容性,且毒副作用小等优势,但是肽类抑制剂相对分子质量较大,生物稳定性差,口服利用率低,且不容易通过血脑屏障[7]。因此,小分子化合物抑制剂成为近年来研究BACE1抑制剂一大热点。与之前的肽类抑制剂相比较,它们具有相对分子质量小、生物代谢活性稳定、膜通透性好等特点。目前,这些非肽类小分子抑制剂一般是通过计算机辅助设计的方法完成的,这种方法大大降低了研发成本,缩短研发周期[8]。本文主要采用分子对接方法,利用AutoDock4.2软件[9]将20种吡咯烷酮类螺环化合物抑制剂对接到了BACE1的活性中心。基于分子对接方法得到的抑制剂在BACE1活性中心的结合构型,分析了影响抑制剂生物活性的主要因素,揭示其与 BACE1 的作用机制,为抑制剂的设计优化和结构改造提供了重要参考。
1 计算方法
1.1 分子对接
人们已经通过实验的方法得到了BACE1的晶体结构,BACE1晶体结构在蛋白质数据库中的PDB编码为3UDH。我们首先从PDB数据库中下载了BACE1的晶体结构3UDH。实验得到的BACE1的晶体结构3UDH的活性中心中含有配体小分子,我们通过Chimera[10]软件将配体小分子从蛋白质的活性中心剥离。然后使用AutoDock4.2软件将晶体结构中的水分子删除,由于实验得到的晶体结构中并不包含氢原子,在AutoDock4.2软件中我们对BACE1的极性基团进行加氢处理。并在AutoDock4.2软件中对蛋白质进行了电荷计算。
本文中所有的分子对接过程均是在AutoDock4.2软件中完成的。20个抑制剂分子与BACE1的分子对接中,所有的对接格点间隔均设为0.375,所构建的对接盒子大小为42×48×54,中心(x,y,z)设置为20.86,35.66,40.68。所有的分子对接过程均采用刚性对接,即在对接模拟中BACE1的蛋白结构保持不变。本文中所有对接均采用拉马克遗传算法(LGA)。在RMSD为2.0Å下对对接结果进行构象聚类收集。
1.2 小分子构建
为了研究小分子抑制剂与BACE1的结合机制,我们基于吡咯烷酮类螺环化合物构建了20个小分子BACE1抑制剂,如表1所示。我们将构建的20个BACE1抑制剂采用分子对接方法对接到了BACE1的活性中心。对接过程中,我们考虑了所构建的BCAE1抑制剂所有可以自由旋转的共价键,使抑制剂分子在对接过程中能够自由旋转的键达到最大值。
2 结果
2.1 BACE1活性中心结构

图1 BACE1及其活性口袋
我们首先使用Chimera软件对BACE1的晶体结构进行了,β-分泌酶的活性口袋由Leu30、Asp32、Ser35、Pro70、Tyr71、Thr72、Gln73、Phe108、Trp115、Ile118、Tyr198、Ile226、Asp228、Thr231、Arg235、Val332等残基构成。
2.2 抑制剂与BACE1之间的结合能
为了考查抑制剂分子与BACE1之间的作用机制,我们使用Autodock4.2分子对接模拟软件,将20种BACE1抑制剂小分子分别对接到了β-分泌酶的活性中心,得到了20种抑制剂分子与BACE1结合的结合能,如表1所示。
表1 抑制剂分子式及与BACE1对接的结合能
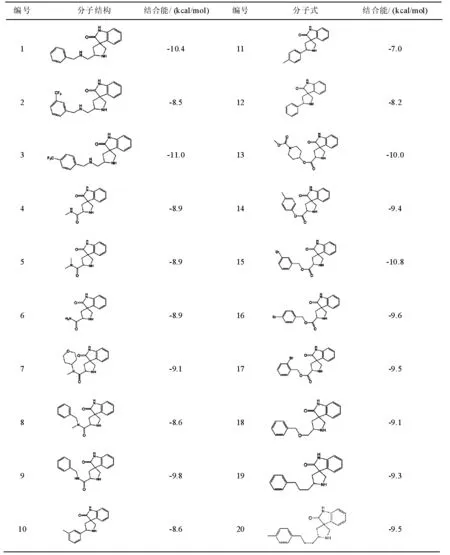
2.3 抑制剂在BACE1活性中心的结合构象
为了考查抑制剂分子在BACE1结合位点的构象,我们将对接结果在均方根偏差RMSD为2.0Å的条件下进行了构象聚类分析,将聚类分析得到的构象最大簇在BACE1的活性口袋进行了构象重叠,如图2所示。此外,为了探究抑制剂分子与BACE1的结合机制,我们选取了每组对接结果中的一个典型构型进行了分析,将每组对接结果的典型构型在BACE1的活性中心显示,如图3所示。
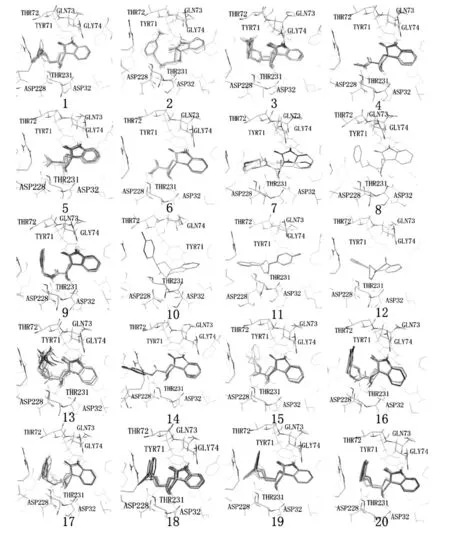
图2 抑制剂小分子在活动中心对接结果重叠图
3 讨论
经Chimera软件分析可知,BACE1活性口袋中的Leu30、Pro70、Thr72、Phe108、Trp115、Ile118、Ile226、Thr231、Val332等疏水性残基构成了β-分泌酶活性口袋的疏水通道。
抑制剂分子与BACE1之间的结合能越大,抑制剂分子越容易与BACE1结合。由表1可知,抑制剂分子1、3、13、15与BACE1的之间的结合能较大,它们分别是-10.4、-11.0、-10.0、-10.8 kcal/mol;说明这几个抑制剂分子能够很好的与BACE1结合,具有很好的生物活性;而抑制剂分子11、12与BACE1的结合能较小,分别为-7.0、-8.2 kcal/mol,说明这几个抑制剂分子与BACE1的结合能力较弱。
由图2可知,20种抑制剂被很好的对接到了BACE1的活性中心,且得到的对接结果都能够很好的重叠。同时,BACE1活性口袋中的Leu30、Pro70、Thr72、Phe108、Trp115、Ile118、Ile226、Thr231、Val332等疏水性残基构成的疏水通道很好的将抑制剂分子中的疏水性基团包裹。小分子中的疏水性基团与活性口袋中的等疏水性残基形成了疏水作用区域,使得受体小分子被包裹在疏水口袋中,从而使小分子被很好的对接到了BACE1的活性口袋中,这种作用有利于构象的稳定。
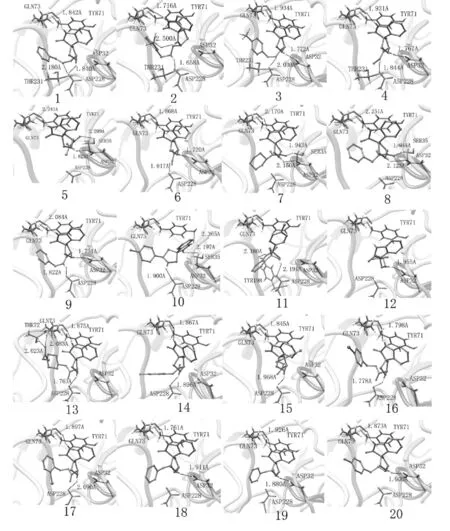
图3 每组最优结果与活性中心对接构象
从图3中可知,20种抑制剂分子母体吡咯烷酮亚氨基上的H与活性中心Gln73上的羰基之间形成了非常强的氢键作用,这是抑制剂能够抑制BACE1活性的一个主要因素。此外,抑制剂母体吡咯环上的亚氨基较容易与Asp32上的羧基形成强的氢键作用。当抑制剂分子取代基上含有氨基或亚氨基时,取代基上的氨基或亚氨基也较容易和Asp228上的羧基之间形成氢键。抑制剂与BACE1活性中心形成的氢键网络将抑制剂分子固定在BACE1的活性中心。这与实验得到的结论一致[11]。
抑制剂分子与β-分泌酶形成的氢键有利于抑制剂小分子与β-分泌酶的结合,降低抑制剂分子与BACE1结合的结合能。由图3可知,抑制剂分子3除了母体中的吡咯烷酮环上的亚氨基与Gln73形成氢键之外,还有另外两个氢键生成,分别是吡咯环上的亚氨基与Asp228生成的氢键,以及抑制剂分子取代基上的亚氨基与活性口袋中的Asp228的羰基形成的氢键。抑制剂分子3与BACE1之间形成了一个非常强的氢键网络,使抑制剂分子3较容易与BACE1结合,这也是抑制剂分子3与BACE1结合能较低的一个关键因素。而抑制剂分子12仅仅只有母体吡咯烷酮、吡咯环上的亚氨基与活性口袋中的Gln73、Asp228形成氢键,抑制剂分子12与BACE1之间形成的氢键数量较少,这也是抑制剂分子12与BACE1之间结合能较高的一个主要因素。
此外,活性口袋Tyr71侧链上的苯基还与抑制剂吡咯烷酮核心结构上的苯基以面对面相互平行的方式朝向,两个苯基之间生成了芳香交互作用,形成了π-π重叠相互作用。π-π堆积作用的形成有利于抑制剂在BACE1活性中心的稳定性,增强吡咯烷酮类螺环化合物抑制剂的生物活性。本文研究结果与实验报道的结果一致,为以后研究以螺吡咯烷酮核心结构为母体、以β-分泌酶为靶标的新型药物开发提供了机理支持及指导。
4 结论
本文利用AutoDock4.2程序研究了吡咯烷酮类螺环化合物抑制剂与BACE1之间的作用机制。发现抑制剂与活性口袋中的Gln73、Asp228、Asp32之间形成了非常强的氢键网络。此外,BACE1活性中心Tyr71侧链上的苯环与抑制剂母体中的苯环形成了π-π堆积作用。π-π堆积作用是吡咯烷酮类螺环化合物与BACE1之间作用的一个典型结构特征。
