基于全基因组重测序技术的‘红叶’杜仲SNP位点开发
2019-11-15陈梦娇杜庆鑫朱景乐杜红岩杨绍彬
杨 赟 陈梦娇 杜庆鑫 朱景乐 杜红岩 杨绍彬*
(1.国家林业和草原局泡桐研究开发中心,经济林种质创新与利用国家林业和草原局重点实验室,郑州 450003; 2.南京林业大学林学院,南京 210000; 3.河南农业大学林学院,郑州 450002)
杜仲(Eucommiaulmoides)隶属于杜仲科(Eucommiaceae),单属单种,染色体2n=2X=34,是我国特有的经济林树种,在我国北纬22°~42°,东经100°~121°的广大范围内均可栽培[1]。杜仲因其适应性强,干型通直,冠型优美,主根较深,侧根发达,具有较好的水土保持能力,被广泛用于园林绿化和防止水土流失[2~3]。当前,对于杜仲的研究主要集中在药理功效、成分分析、栽培模式,干旱胁迫响应等方面[4~6],对选育观赏型杜仲良种报道较少。‘红叶’杜仲(Eucommiaulmoides‘Hongye’)是中国林业科学研究院选育的观赏型良种,叶片鲜红至紫红色,是理想的绿化树种。‘红叶’杜仲叶片呈红色的主要物质是花色苷[7],其叶片花色苷含量显著高于普通杜仲,但是‘红叶’杜仲叶片的呈色机理尚不清楚。
随着测序技术的不断发展,拥有参考基因组的物种越来越多,全基因组重测序技术在发掘与植物表型相关的单核苷酸多态性(SNP)位点方面发挥着重要作用。研究者利用该技术,快速得到这一物种某个体的基因组信息和重要功能基因的序列信息,初步定位和鉴定差异位点,为从分子水平研究植物基因调控提供了依据[8~10]。植物叶片的呈红色的物质是花色苷,花色苷代谢途径是目前研究的最透彻的通路之一。花色苷由苯丙氨酸经过一系列的酶催化合成,由上游基因群和下游基因群控制酶的合成,上游基因群通常编码参与黄酮、黄酮醇和糅红合成相关的酶,下游基因群编码合成花青苷和原花青素的酶[11]。本研究通过重测序技术检测并分析控制‘红叶’杜仲与普通绿叶杜仲叶色差异的基因位点,比较与叶色形成相关的酶基因位点差异,开发杜仲红叶性状SNP标记。
1 研究方法
1.1 研究材料
‘红叶’杜仲(Eucommiaulmoides‘Hongye’)和‘小叶’杜仲(Eucommiaulmoides‘Xiaoye’)均是在河南洛阳地区通过实生选育方法获得的无性系良种。本研究以3年生‘红叶’杜仲和‘小叶’杜仲嫁接苗为对象(砧木均为普通杜仲),每个无性系选择3个单株,每个单株选择顶端中上部的3片叶片,按无性系将叶片分组后置于液氮中保存。
1.2 DNA提取
按照Qiagen公司的DNeasy Plant Mini Kit试剂盒的操作步骤提取杜仲DNA,使用Nano Drop 2000进行DNA纯度检测,Picogreen方法检测DNA浓度。1%的琼脂糖凝胶电泳检测DNA完整性,检测前将样品放在冰上融化后,充分混匀并离心。每个无性系3个单株,从中取等量的质量较好的DNA混合建库,用于二代测序。
1.3 全基因组重测序
将检验合格的基因组DNA样品(样品量>2 μg,样品浓度>50 ng·μL-1,无色素污染,无明显RNA、蛋白质等杂质污染),送至上海美吉生物医药科技有限公司进行文库构建。利用超声波将DNA序列片段形成随机片段,经过处理后用磁珠吸附富集基因组长度为400 bp左右的片段,经过PCR扩增形成测序文库。质检合格的文库用Illumina HiSeqTM平台进行基因组重测序,测序策略为Illumina PE150,总测序读长为300 bp。
1.3.1 原始测序数据质控与过滤
原始测序数据(Raw Data)经过Fastp(v0.11.5)进行质控,包括碱基含量分布和碱基错误率分布。碱基含量分布检查有无AT、GC分离现象。测序碱基序列错误率结果由于Illumina HiseqTM测序的技术特点,测序片段前端几个Cycles和末端的错误率会偏高。使用NGSQCtoolkit(v2.3.3)套件对raw reads去除接头等对reads进行trimming,去除低质量read并剪切掉5′端及3′端3 bp碱基并过滤掉长度小于50 bp的reads。最终得到质量较好的Clean Data。测序数据过滤采用Fastp默认参数流程[12]。
1.3.2 变异位点检测及注释
对于‘红叶’杜仲和‘小叶’杜仲测序获得的Clean Data,利用BWA(v0.7.16)基于MEM算法[13]将其比对到杜仲参考基因组序列上(杜仲参考基因组叶片为绿色),利用GATK(v3.8.0)对BAM文件进行校正[14],并进行SNP检测。利用GATK软件的Haplotyper算法进行变异位点检测。利用SnpEff(v4.3)进行SNP变异位点功能注释[15],包括变异位点位置和功能注释;SnpEff会根据变异位点对蛋白编码的影响,从高到低依次分为High、Moderate、Low和Modifier 4个等级,High代表严重影响功能的SNP个数,Moderate代表中度影响功能突变的SNP;Low代表低影响程度;Modifier表示与功能无关的SNP。因此,只关注High和Moderate的个数。
1.3.3 变异基因的功能注释和代谢通路分析
利用Diamond软件比对到NR、UniProt和Gene Ontology(GO)数据库,进行基因功能预测。使用KOBAS软件比对到KEGG数据库,注释基因所涉及的代谢通路。利用emappe软件比对EggNog数据库,注释基因功能。
1.3.4 变异位点的筛选
首先根据变异位点对蛋白编码的影响,构建导致蛋白发生High、Moderate变化的基因集。根据变异基因的功能注释,筛选与类黄酮和花色苷合成途径相关的基因集。为了保证SNP位点的准确性,过滤掉低质量的SNP位点,要求SNP深度≥10X。选取‘红叶’杜仲与参考基因组完全不一致的纯合位点,‘小叶’杜仲与参考基因组完全一致的纯合位点,这些位点被用于SNP位点开发。
1.4 基于Sanger测序SNP验证
参考杜仲基因组序列信息,采用Primer Premier 5设计引物,退火温度在60°左右,引物长度18~30 bp。PCR扩增产物长度:外显检测引物离外显子上下游150 bp左右;目的基因测序的PCR产物条带一般不超过1 200 bp。PCR反应体系:50 μL:1~2 μL的50 ng·μL-1cDNA,上下游引物、dNTP(mix)均2 μL(10 mmol·L-1), 10×Taq Buffer(with MgCl2) 5 μL,Taq酶(5 U·μL-1)0.5 μL,加双蒸水至50 μL。PCR反应条件:95℃预变性3 min; 然后94℃变性30 s,60℃退火30 s,72℃延伸90 s,共35个循环;最后72℃延伸8 min,4℃结束。Sanger测序由上海生工生物工程公司完成。
2 结果与分析
2.1 原始测序数据统计及质控
本次测序质量较好。‘红叶’杜仲获得14.16 G数据量,47 032 423个Clean reads,其中碱基质量Q30比例达到91.72%,GC含量为35.54%。‘小叶’杜仲获得14.29 G数据量,47 476 746个Clean reads,其中碱基质量Q30比例达到91.93%,GC含量为35.43%。数据质控分析显示,除了reads的前几个碱基存在正常的不平衡现象外,碱基质量分布基本无AT、GC分离现象,测序碱基错误率分布统计低于0.001%。
2.2 变异位点检测结果
2.2.1 比对参考基因组数据统计
杜仲参考基因组大小为1.23 Gb,组装水平是Scafflod(Scafflod N50=1.88 Mb)。比对参考基因组,‘红叶’杜仲和‘小叶’杜仲测序数据的Mapped Ratio均为99.90%。本研究两样本对参考基因组(排除N区)的平均覆盖深度为10×左右,1×覆盖深度(至少有一个碱基的覆盖度)为87.77%以上。本研究覆盖到参考基因组的区域较多,可检测到的变异位点较多,基因组上碱基的覆盖深度分布较均匀,测序的随机性好。
2.2.2 SNP的检测
在‘小叶’杜仲中共检测到5 711 141个SNP,其中发生转换(Ti)的SNP有4 139 309个,发生颠换(Tv)的SNP有1 565 071个,Ti/Tv比值为2.64,在‘红叶’杜仲中共检测到5 407 823个SNP,其中发生转换(Ti)的SNP有3 915 050个,发生颠换(Tv)的SNP有1 487 022个,Ti/Tv比值为2.63。
2.2.3 SNP差异位点对蛋白编码影响的评估
在‘小叶’杜仲中存在严重影响蛋白质功能的SNP为1 640个,中度影响功能的SNP为47 192个。在‘红叶’杜仲中存在严重影响功能的有1 516个SNP,含有中度影响的41 328个SNP。
2.2.4 SNP差异位点功能注释
SnpEff每个变异类型的功能统计如表1所示。某一基因在‘红叶’杜仲和‘小叶’杜仲间存在碱基差异。‘红叶’杜仲和‘小叶’杜仲每个SNP类型在数量上差异不大,可能它们分布在不同基因上,导致了‘红叶’杜仲和‘小叶’杜仲在表型上的差异。
2.3 变异基因的功能分类
2.3.1 变异基因的NR、Uniprot、GO、KEGG、EggNOG注释
含有变异位点的26 722基因,在Nr数据库注释到13 408条基因,在Uniport中注释到14 418条基因,在GO数据库中注释到21 453条基因,在EggNOG数据库中注释22 660条基因,在KEGG数据库中注释到5 915条基因。为了进一步研究含有突变位点的基因功能,向GO数据库的各个term进行映射(图1),发现在细胞组分(cellular component)中注释到核、细胞质、膜的有机组成部分,在生物进程(biological process)中注释到等离子体膜,在分子功能(molecular function)中注释到蛋白结合、线粒体和叶绿体、ATP结合及转录过程。根据KEGG注释信息,如表2所示,含有差异位点的基因可注释到286条代谢途径,其中较多的是淀粉和蔗糖代谢,苯丙素生物合成途径等,与植物叶片生长及颜色密切相关。利用EggNOG数据库对比分析,如图2所示,有12 977个位点注释到25个类别,其中信号转导机制(1 220,9.38%)、转录(868,6.68%)、蛋白质翻译后修饰与转运、分子伴侣(838,6.45%)碳水化合物的运输和代谢(801,6.16%)等类别注释到的变异位点多,其次是能量产生和转换(527,4.05%),次生代谢产物的生物合成、转运和分解代谢(456,3.51%)。

图1 GO功能富集分类前20位统计图 横坐标表示位于功能区属于High和Moderate类型的变异位点的个数,纵坐标表示GO Term的ID。Fig.1 GO function enrichment classification chart The abscissa indicates the number of variant sites located in the functional area belonging to the High and Moderate types,and the ordinate indicates the ID of the GO Term.
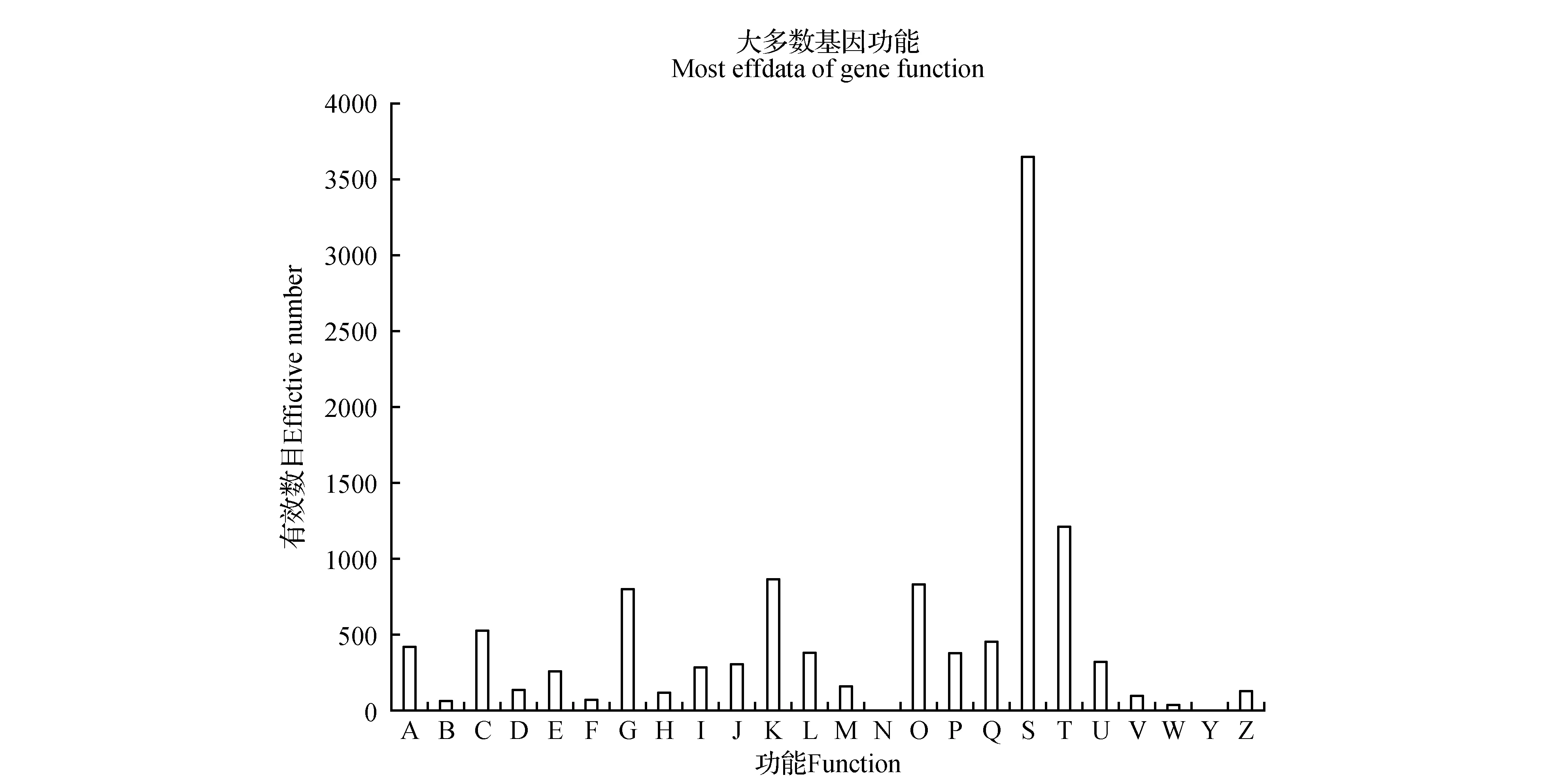
图2 COG功能分类图 A. RNA加工和修饰;B.染色质结构和动力学;C.能源生产和转换;D.细胞周期控制,细胞分裂,染色体分配;E.氨基酸转运和代谢;F.核苷酸转运和代谢;G.碳水化合物转运和代谢;H.辅酶转运和代谢;I.脂质转运和代谢;J.翻译,核糖体结构和生物发生;K.转录;L.复制,重组和修复;M.细胞壁/膜/包膜生物发生;N.细胞运动性;O.翻译后修饰,蛋白质周转,分子伴侣;P.无机离子转运和代谢;Q.次生代谢产物的生物合成,分解代谢;S.功能未知;T.信号转导机制;U.细胞内运输,分泌和囊泡运输;V.防御机制;W.细胞外结构;Y.核结构;Z.细胞骨架Fig.2 COG function classification chart A. RNA processing and modification; B. Chromatin structure and dynamics; C. Energy production and conversion; D. Cell cycle control,cell division,chromosome partitioning; E. Amino acid transport and metabolism; F. Nucleotide transport and metabolism; G. Carbohydrate transport and metabolism; H. Coenzyme transport and metabolism; I. Lipid transport and metabolism; J. Translation,ribosomal structure and biogenesis; K. Transcription; L. Replication,recombination and repair; M. Cell wall/membrane/envelope biogenesis; N. Cell motility; O. Posttranslational modification,protein turnover,chaperones; P. Inorganic ion transport and metabolism; Q. Secondary metabolites biosynthesis,and catabolism; S. Function unknown; T. Signal transduction mechanisms; U. Intracellular trafficking,secretion,and vesicular transport; V. Defense mechanisms; W. Extracellular structures; Y. Nuclear structure; Z. Cytoskeleton
表1 SNP突变位点功能注释统计表
Table 1 SNP mutation site function annotation statistics table

类型Type‘红叶’杜仲E.ulmoides‘Hongye’‘小叶’杜仲E.ulmoides‘Xiaoye’5′ 端非翻译区5_prime_UTR_variant53下游Downstream707591753545基因间区Intergenic region48268425097746内含子区Intron variant503340532407外显子区域的错义突变Missense variant4132843402非编码转录本区Non coding transcript variant1810内含子左侧连接区Splice acceptor variant188197内含子右侧连接区Splice donor variant124149距离内含子或外显子2bp的剪切区Splice region variant55275952突变导致启动子缺失Start lost165181突变导致终止子获得Stop gained753807突变导致终止子突变Stop lost287307突变导致终止子的编码改变Stop retained variant8288同义突变Synonymous variant4051742898上游Upstream gene variant759898809593
2.3.4 用于分子标记开发的SNP位点筛选
根据基因功能注释的5大数据库结果,26 722条含有差异位点的基因中共有228条基因注释到与花色苷和类黄酮合成相关。228条基因内部共有829个差异位点,根据SnpEff注释,46个位点严重影响蛋白质结构,783个位点中度影响蛋白质功能。与参考基因组比,‘红叶’杜仲中存在的这228条基因,含有27个位点严重影响蛋白质结构,其中有15个纯合位点;有556个位点中度影响蛋白质结构,269个纯合位点。经过最终筛选,得到12个位点在‘红叶’杜仲中完全差异于参考基因组,且‘小叶’杜仲中的这12个位点与参考基因组一致,测序深度均≥10×(表3)。这12个位点分布在6条基因的内部(表4)。
表2 杜仲重测序的KEGG代谢途径分类图
Table 2 Classification of KEGG metabolic pathways forEucommiaulmoidesresequencing

编号Number途径PathwayDifferent sites with KEGG annotation(12997)ko ID1淀粉和蔗糖代谢Starch and sucrose metabolism183(1.41%)ko005002植物—病原互作Plant-pathogen interaction164(1.26%)ko046263苯丙素的生物合成Phenylpropanoid biosynthesis128(0.98%)ko009404碳代谢Carbon metabolism125(0.96%)ko012005植物激素信号转导Plant hormone signal transduction105(0.81%)ko040756氨基酸的生物合成Biosynthesis of amino acids103(0.79%)ko012307核糖体Ribosome98(0.75%)ko030108内质网蛋白加工Protein processing in endoplasmic reticulum98(0.75%)ko041419戊糖和葡萄糖醛酸酯互变Pentose and glucuronate interconversions92(0.71%)ko0004010氨基糖和核苷酸糖代谢Amino sugar and nucleotide sugar metabolism92(0.71%)ko0052011RNA运输RNA transport73(0.56%)ko0301312糖酵解和糖质新生Glycolysis/Gluconeogenesis72(0.55%)ko0001013嘌呤代谢Purine metabolism67(0.52%)ko0023014剪接体Spliceosome64(0.49%)ko0304015真核生物核糖体生物起源Ribosome biogenesis in eukaryotes60(0.46%)ko0300816半乳糖代谢Galactose metabolism59(0.45%)ko0005217内吞作用Endocytosis59(0.45%)ko0414418氰氨基酸代谢Cyanoamino acid metabolism57(0.44%)ko0046019细胞周期Cell cycle55(0.42%)ko0411020抗坏血酸和新陈代谢Ascorbate and aldarate metabolism54(0.42%)ko00053

表3 12个与红叶性状相关的SNP位点

表4 筛选出的6条基因的功能注释

表5 SNP的验证结果

图3 基于SNP标记的红叶性状的初步分子鉴定Fig.3 Preliminary molecular identification of red leaf traits by SNP markers
2.4 Sanger测序结果
基于重测序数据和SNP位置设计了6对引物,一代测序的验证结果与重测序筛选结果,匹配率100%。共筛选出12个分子标记用于‘红叶’杜仲红叶性状的早期鉴定(表5)。图3给出了表5中序号1和2的SNP验证结果。
3 讨论
杜仲是单科单属单种,‘红叶’杜仲和‘小叶’杜仲均是在河南洛阳地区通过实生选育得到的无性系良种,遗传背景相对较小。本研究在参考杜仲基因组大小(1.18 G)的基础上,前期按10×的测序深度,后期在分析中设定严格的阈值,同时再进行部分sanger测序验证,以期尽可能准确地得到SNP位点。根据重测序结果,在‘红叶’杜仲中共检测到5 407 823个SNP,583个位点能够严重或者中度影响蛋白质结构,且与花色苷合成相关。目前也有一些学者采用同样的方法得到类似的结论,如Ni等[16]对日本杏红绿皮果实2个品种的全基因组重测序分析,对CDS区域6个序列数据集的比对分析发现181 331个SNPs,3 318个InDels和51 427个SVs,筛选出与红色果皮颜色形成相关的13个候选基因。Xu等[17]对两种栽培葡萄株系的幼苗叶片进行基因组重测序,筛选出635个sbc特异性基因和665个sbbm特异性基因,分析了非同义突变与控制颜色形成的基因的关系,发现NCED6表达与NCED1有关,并可能影响花青素积累。此次研究可能存在一些未覆盖的位点,得到的数据准确性也可能有一定偏差,今后我们会加大群体和测序规模,继续深入开展相关研究。
‘红叶’杜仲和‘小叶’杜仲除了叶片颜色外叶形也存在一定差异,我们在分析中锁定的是与花色苷合成相关的代谢通路,以尽量避免其他因素的干扰。目前多种研究方法共同揭示花色苷代谢途径中SNP位点差异被广泛使用。Jiao等[18]在14种中国野生葡萄品种的样本中克隆了调节花色素苷生物合成的mybA的两个转录因子VvmybA1和VlmybA2,在测序的片段中检测到总共121个SNP,这些SNPs分布在启动子区、内含子区和编码区。Liu等[19]利用MISA、GATK3和注释工具,根据转录组数据从毛茛花瓣151,136个单基因中,鉴定和定位SSR和SNP标记。在8个花青素合成关键酶基因家族的116个单基因中,鉴定出127个SNP。ANS酶基因中没有SNP位点,而DFR酶基因具有最大量的SNP位点。Oh等[20]利用全基因组重测序检测SNP和转录组联合分析黑米花青素的合成调控,发掘到9个候选基因中的3个基因与花色苷的合成有很好的相关性,尤其是基因Os06t0192100(UDP糖基转移酶)与之前报道的Os04g0557500基因相似性很高。这样联合分析的方法为我们进一步深入研究‘红叶’杜仲花青素的系统进化和基因通路提供了思路。在本研究结合‘小叶’杜仲的重测序结果与一代测序,发现12个纯合差异位点是‘红叶’杜仲特有的,更验证了12个与‘红叶’杜仲红叶性状相关的SNP位点。下一步将选择叶色有差别的杜仲,通过PCR扩增测序的方法对上述SNP位点开展进一步验证。
导致‘红叶’杜仲叶片特殊呈色的原因可能会有很多,如基因变异、基因表达量的差异及表观遗传修饰的不同等,本研究仅从基因层面通过SNP位点差异初步探讨‘红叶’杜仲呈现红色的可能原因,至于确切的原因,还需进一步的探索。
