金属单原子模型催化剂热稳定性的反应力场(ReaxFF)分子动力学研究
2019-11-08杨文琦乔园园王贵昌
杨文琦 汪 杰 乔园园 王贵昌
(南开大学化学学院,先进能源材料化学教育部重点实验室,化学科学与工程协同创新中心(天津),天津 300071)
单原子催化剂(SAC)在催化领域是相对较新的概念,在过去几年中受到人们的广泛关注[1-8]。由于它具有很高的化学势,金属单原子很容易迁移并聚集形成大的纳米颗粒。如果一个金属单原子想在高温时稳定存在,那么就要求单原子和金属基底之间具有强相互作用。较强的相互作用可以阻碍单个原子凝聚烧结,从而保持稳定。如果这个相互作用过于强,催化剂会失去它的催化活性。因此平衡催化剂活性和稳定性这两者的关系是很有必要的。单原子催化剂烧结过程包括以下几步:首先原子从基底表面脱离成为小的纳米颗粒,之后发生迁移从而形成较大的纳米颗粒,也就会发生凝聚烧结现象[9]。一般而言,SAC催化剂往往是过渡金属负载在过渡金属氧化物上,金属-载体的强相互作用有利于SAC的稳定,如Datye课题组[6]发现氧化铈与铂的相互作用可以稳定氧化铈部分晶面上孤立的Pt原子,可以在高温时保持分散的Pt原子不聚集。另一方面,对于高度配位不饱和的过渡金属单原子催化剂(如adatom)则研究的还不够。
Kentaro等通过场离子显微镜研究了Pt(111)上Pt团簇的形成以及迁移能垒与温度的关系,发现在350 K的低温下,包括五聚体在内的Pt团簇扩散和解离现象都是比较显著的[10]。Kokalj及其合作者[11]发现Rh1/Rh(111)-ad-tom催化剂可以增强甲烷第一次脱氢形成甲基的反应,同时可以抑制甲基的进一步脱氢反应,从而既保证了甲烷的充分活化,同时也避免了积碳现象的发生。那么,该类ad-atom催化剂在反应温度下是否可以稳定存在呢?为探讨该问题,在本工作中,我们通过反应力场(ReaxFF)结合LAMMPS(large-scale atomic/molecular massively parallel simulator)软件包进行了大尺度分子动力学模拟,研究了一系列ad-atom过渡金属催化剂(Cu,Ag,Au,Fe,Ni,Pd,Pt,Ru)在高温的稳定性。 同时,通过与密度泛函(DFT)计算结果进行对比,验证Ni/C/H/O力场参数,并探究了金属单原子催化剂模型Ni1/Ni(111)在H2和O2气氛下的稳定性行为。该研究工作为人们制备高活性和选择性ad-atom催化剂提供了一定的理论指导依据。
1 计算方法
采用基于密度泛函理论的从头量子力学的程序包VASP(Vienna ab-initio simulation package)[12-13]进行计算。基组采用平面波赝势方法[14-15],利用PBE关联梯度修正泛函对模型进行充分优化[16],采用广义梯度近似(GGA)描述体系的交换关联能。平面波的截断能设置为 400 eV,k 点[17]设置为 3×3×1。 对于含有Fe和Ni的体系,加入了自旋极化。晶胞厚度为4层,扩胞3×3,单原子催化剂模型(M1/M(111))为添加一个额外的原子在fcc或hcp位点。在优化过程中,上3层驰豫,下2层原子保持固定。
大尺度分子动力学模拟软件包LAMMPS[18]和反应性力场(ReaxFF)用于分子动力学的计算。动力学模拟所用模型为Cu单胞切(111)表面,厚度为4层,扩9×9超胞的周期性slab模型。这4层中下2层固定,上面2层放开,在表层上方有1.5 nm的真空层。时间步长设为0.25 fs,模拟时间设为100 ps,系综选择NVT系综。首先设置初始温度100 K模拟10 ps稳定,然后用50 ps将系统升温至所需温度(最终温度为 300,400,500和 600 K), 温度阻尼为 25.0,最后模拟100 ps来稳定此状态。SAC体系(Ag,Au,Fe,Ni,Pd,Pt,Ru 和 Cu) 反 应 性 力 场 参 数 来 源 于Duin等的工作[19-28]。Ni/C/H/O的力场参数基于Ni/C/H和Pt/O的力场参数[23,25]。
2 结果与讨论
2.1 验证反应力场参数的可靠性
我们首先对力场参数的可信度进行了验证。在训练集中加入了Ni块体(bulk)的状态方程,金属Ni的内聚能(Ecohesive,见式1,EMbulk表示金属M块体的能量,NMbulk表示块体中包含有金属M原子的数目,EMatom代表气相 中 金 属 M 原子的能量),CH3,CH2,CH,H2和O2等吸附物种在Ni(111)表面上的结合能(Ebinding,见式 2,Esp/Ni(111)和 ENi(111)分别代表吸附物种吸附后的体系和吸附前Ni(111)slab的能量,Espatom表示吸附物种在气相中的能量),Ni(111)面的表面能(Esurf,见式 3,Eslab,EMbulk和 NM分别为 Ni(111)slab 模型的能量,块体中金属Ni原子的能量和slab模型中Ni原子数目,A是切面的面积),以及H2和O2在载体上分解的反应热。采用单参数搜索法[29],优化了Ni和H,Ni和O的键参数,以及Ni和H,O的交叉项。

表1 DFT计算结果和ReaxFF拟合的H 2和O2分别在Ni(111)和Ni1/Ni(111)上分解的反应热的比较Tabel 1 ReaxFF fits for the reaction energy of the dissociation of H 2 and O2 on Ni(111)and Ni/Ni(111)respectively kJ·mol-1

图1 不同吸附物种吸附在Ni(111)不同位点以及H2和O2吸附在Ni1/Ni(111)top位点上DFT和反应力场拟合的吸附能结果Fig.1 ReaxFFfits for the binding energy of different adsorbed species(CH3,CH2,CH,O and H)on the different site of Ni(111)surface and H2,O2 molecular on the Ni1/Ni(111)

图1和表1结果表明力场参数对所研究体系吸附能和反应热进行了拟合,说明本文所用力场参数是可靠的。
2.2 单原子金属催化剂(M 1/M(111),M=Cu,Ag,Au,Pd,Pt,Ni,Ru,Fe和Fe1/Fe(100))的稳定性

图2 从上至下分别为Cu,Ag,Au,Ni,Pd,Pt,Ru,Fe单金属M1/M(111)模型和Fe1/Fe(100)模型在300,400,500和600 K时动力学构型(100 ps)的侧视图Fig.2 Side view of dynamics configurations(100 ps)Cu,Ag,Au,Ni,Pd,Pt,Ru,Fe single metal model from top to bottom respectively and Fe1/Fe(100)model at 300,400,500 and 600 K
对于单原子金属催化剂模型,我们使用分子动力学方法研究了具有分散的单原子表面在不同温度下的热稳定性。通过DFT对四层扩胞3×3的Cu1/Cu(111)进行结构优化,然后将优化后的构型再次扩胞3×3最终形成一个四层扩胞9×9的结构,在nvt系综100 K的低温下模拟25 ps达到平衡。模拟温度从100 K逐步升温至300 K,并在300 K温度下模拟100 ps,对模拟三次后的结果进行统计。发现单金属原子Cu催化剂会发生聚集(图2),并且随着温度增加聚集的原子数增加。例如在300 K时有4~5个原子聚集形成五聚体,而400~500 K时9个Cu原子发生聚集,甚至在600 K时表层原子排列变得更加混乱,Cu原子显著驰豫。这表明单原子Cu在高于300 K时不能稳定存在。
对于大部分过渡金属 (如 Ag,Au,Pd,Pt和 Ni),随着温度升高,单金属原子模型变得不稳定并聚集在一起形成金属团簇。有趣的是对于Fe1/Fe(100)模型,它可以稳定地存在不产生团簇(图2),但Fe1/Fe(111)模型的结构不能保持稳定。这说明金属表面结构和其原子的排列也是影响单原子金属催化剂稳定性的重要因素。如图3所示,通过DFT计算可知大多数过渡金属(除了Fe)均具有内聚能(Ecohesive)高于结合能(Ebinding)的性质(计算方法见公式(1)和公式(2)),说明单原子的聚集是热力学有利的过程,因此倾向于聚集。然而对于铁,它的内聚能小于结合能,单金属铁原子可以被有效地束缚住,意味着聚集过程不是热力学有利过程,因此Fe单原子可以稳定地存在。

图3 金属单原子和载体的吸附强度以及对应的金属原子的聚合能两者之间的关系Fig.3 Relationship between binding strength of single atom and the corresponding cohesive energy
高温时大部分ad-atom催化剂不稳定,因此研究低温时M1/M(111)表面单原子团聚成簇现象与温度的关系(如图4所示),同样温度下团簇中原子数越少越稳定。可以看出低于200 K稳定性Au(Ru)>Pt>Cu>Pd>Ag>Ni,300 K 时 Au>Ru(Cu)>Pt(Pd)>Ag(Ni)。而Pt1/Pt(111)在实验中160 K时形成二聚体,210 K时形成三聚体[10],与本工作中模拟结果(165 K时形成二聚体,200 K形成三聚体)比较接近。
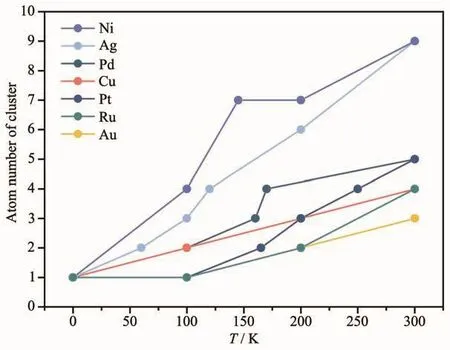
图4 M1/M(111)模型低温时表面单原子发生团聚现象,温度与团簇原子数的关系Fig.4 Relationship between temperature and atom number of cluster when the aggregation of M1/M(111)model accurs at low temperature
2.3 Ni1/Ni(111)单原子金属模型催化剂在H 2(O2)气氛下的稳定性研究
在实验上,催化剂一般是在各种气氛下参与反应,如Ni可以高效地使甲烷部分氧化。因此我们也研究了H2和O2气氛是如何影响模型催化剂(Ni1/Ni(111))的稳定性。100 K 时(图 5),Ni1/Ni(111)催化剂在H2气氛和O2气氛下保持稳定,在真空环境中Ni单原子则会聚集。在H2气氛下,对Ni1/Ni(111)进行动力学研究。发现100 K的模拟温度下,时间进行至9 ps时出现第1个H2分子分解(图S1),至24 ps时出现第2个H2分子分解,之后则无明显变化,并且Ni1/Ni(111)模型催化剂保持稳定。300 K条件时,有更多的H2分解,同时催化剂无法保持稳定,表面孤立的Ni单原子团聚成簇。在O2气氛下,负载的Ni单原子移动受到O2分子阻碍,在100 K时保持稳定。当模拟温度设定为300 K时,升温过程中出现O2分子向邻近Ni原子移动的情况(图S1d),持续升温会出现O2分解现象,随后Ni单原子发生聚集。体系升至300 K,全部Ni单原子团聚成簇。而随着温度继续升高至400 K以上,表面Ni原子会脱出表面,分解的少部分O原子则会嵌入表面空穴中。与真空状态模拟结果相比较(图S2),H2(尤其是O2)的存在,一定程度上提高了Ni1/Ni(111)的热稳定性。其原因可能是Ni与O2(或O)之间强的相互作用阻碍了聚合现象的发生。

图5 在H2(a)和O2气氛(b)下,Ni1/Ni(111)催化模型在100、300、400、500和600 K时的动力学模拟结果Fig.5 Dynamics results of Ni1/Ni(111)catalyst model under H2(a)and O2(b)atomosphere at 100,300,400,500 and 600 K
3 结 论
在本工作中,通过反应性力场分子动力学模拟研究了金属载体的单原子催化剂(M1/M(111))在300到600 K的热稳定性。模拟结果表明,对于SAC模型,只有Fe1/Fe(100)催化剂体系可以在高的反应温度(约600 K)中稳定存在,而别的单原子金属催化剂会在载体表面形成金属团簇。原因总结如下:Fe1/Fe(100)的吸附有较强的结合能,高于它的内聚能,而别的过渡金属相反,与载体的结合能力要弱于内聚能,因此Fe1/Fe(100)可以稳定存在。对于Ni1/Ni(111)催化剂,H2和O2气氛在一定程度上提高了它的稳定性,气氛会阻碍Ni单原子在表面发生团聚的过程。
Supporting information is availableat http://www.wjhxxb.cn
