多酸基主客体框架材料POMs@MOFs(COFs)
2019-11-08李季坤赵帅恒胡长文
李季坤 赵帅恒 胡长文*,
(1泰山学院化学化工学院,泰安 271021)(2北京理工大学化学与化工学院,原子分子簇科学教育部重点实验室,光电转换材料北京市重点实验室,北京 100081)
0 引 言
多酸,即多金属氧酸盐(polyoxometalates,简写为POMs),是一类具有纳米尺寸、结构明确的过渡金属氧簇,表现出较高的热稳定性和酸碱稳定性[1-3]。POMs材料有良好的电子储存和氧化还原能力,在催化、光电磁、质子传导及荧光等领域具有广泛的潜在应用前景[4]。特别是在催化领域,自20世纪70年代以来,POMs作为烯烃水合等若干个重要工业过程的绿色催化剂,已实现大规模产业化应用,产生了举世瞩目的影响[5-7]。近年来,POMs在催化光/电驱动分解水产氢产氧[8-10]、石油脱硫[11-12]、化学战剂分解/降解[13-15]以及高附加值化学品制备[16-20]等领域的研究也取得较大进展。然而,在实际催化反应中,POMs材料在均相体系中表现出较高的溶解度、难以再生利用且易于团聚,而在非均相体系中往往具有较低的比表面积和较差的稳定性从而影响其催化活性,这些问题极大地限制了其工业化应用。POMs固载为实现该类催化剂的大规模工业化应用提供了有效途径。POMs的传统固载方法有抗衡离子调控法[21]和以介孔分子筛[22]、水滑石[23]或碳材料[24]为载体的负载法等。这些方法仍存在POMs分散度不高、易于团聚或溶出、难以循环利用等问题。以上策略的局限性导致此类POMs催化剂材料难以满足工业规模化应用的要求。
金属-有机框架(metal organic frameworks,简写为MOFs)材料自Yaghi课题组于1999年首次报道以来[25],化学家们对各种MOFs材料开展了广泛的研究,目前已开发出九万多种MOFs材料。MOFs作为一类结构明确的晶态多孔材料,可以定向设计合成,在非均相催化、均相催化剂载体以及气体贮存、分子分离、药物缓释等众多领域展现出广阔的应用前景[26-28]。近年来,共价有机框架(covalent organic frameworks,简写为COFs)材料的合成及在催化等领域的应用研究也得到了快速的发展[29]。而当MOFs(COFs)作为装载主体时可选择性结合不同的功能性客体分子,达到优化结构、提高性能的目的。其中,将POMs作为活性客体分子引入到MOFs(COFs)介孔中,已成为构筑新型POMs@MOFs(COFs)功能化多孔材料的一个有效途径。
1 POMs@MOFs(COFs)研究概述
POMs@MOFs(COFs)材料是将活性的POMs分子以客体形式利用不同的合成方法封装进MOFs(COFs)介孔笼中,制备得到的系列具有明确组成和结构的多酸基主客体框架材料。该类型多孔材料就结构而言,POMs根据MOFs(COFs)介孔的不同尺寸有选择地分布其中,实现了POMs分子的高度分散,符合绿色化学的原子经济性原则。POMs客体分子的引入,既能得到新颖的结构,又能在保持材料多孔性的同时实现与MOFs(COFs)的功能增效,发挥“1+1>2”的效应,例如,在具有重要应用前景的酸/碱催化领域,POMs@MOFs(COFs)材料可以发挥POMs的Brønsted 酸/碱与 MOFs(COFs)的 Lewis 酸/碱的协同催化效应,增强其催化性能。
由表1可以看出,POMs@MOFs(COFs)型多酸基主客体框架材料的装载主体主要集中在HKUST(Hong Kong University of Science and Technology)、MIL(Materials from Institute Lavoisier)、ZIFs(Zeolitic Imidazolate Frameworks)、UiO(University of Oslo)和NU(Northwestern University)以及COFs等材料,且在催化、光电传感、质子传导以及电极修饰材料等领域的应用研究也取得了较好的进展[30]。本文按照合成方法对近年来制备的多酸基主客体框架材料POMs@MOFs(COFs)进行分类总结,并重点讨论了其性能和应用。

表1 POMs@MOFs(COFs)材料的合成方法及性能和应用Table 1 Synthetic methods,properties and application of POMs@MOFs(COFs)materials

续表1

续表1

续表1
2 POMs@MOFs(COFs)材料的制备方法
POMs作为重要的活性分子与多孔的MOFs(COFs)形成封装型材料的合成及性能和应用研究引起了人们的极大兴趣。如图1所示,POMs@MOFs(COFs)材料最为常见的合成方法有原位合成法、浸渍合成法和机械研磨合成法等。这3种合成方法各有优点,也有局限性,下面分类予以详细介绍。
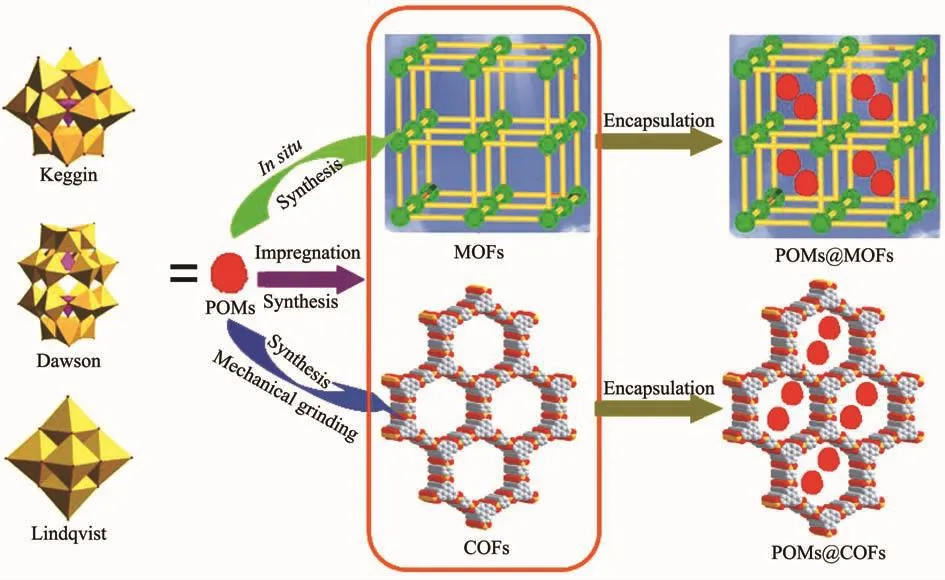
图1 POMs@MOFs(COFs)材料合成方法示意图Fig.1 Schematic representation for the synthetic methods of POMs@MOFs(COFs)materials
2.1 原位合成法
原位合成法又称“瓶中造船法”,该方法是指在MOFs(COFs)的制备过程中加入POMs或合成POMs的初始原料,通过调控反应条件,如溶液pH值、POMs添加量以及选择合适的修饰剂等将POMs一步封装到MOFs(COFs)的孔道结构中。这种方法优点是操作简便,适合于窗口尺寸较小、空腔体积较大的MOFs(COFs)封装POMs材料的制备,但缺点是有些MOFs(COFs)的合成需要较高的温度,甚至会用到一些有机溶剂,这些苛刻的条件使得某些POMs不易保持结构的稳定。而且制备的材料POMs的负载往往不太均匀,故此种方法只适合制备结构稳定的POMs 装载型 POMs@MOFs(COFs)材料[31-73]。
2003年,Naruke课题组用 Cu(NO3)2·3H2O 分别与 Na2MoO4/Na2SiO3、Na2WO4/NaH2PO4和 Na2MoO4/Na2WO4/NaH2PO4体系在四甲基氢氧化铵((CH3)4NOH)、1,3,5-苯三甲酸(H3BTC)存在下原位水热反应首次在HKUST-1骨架中引入POMs,合成了新型纳米 多 孔 化 合 物 SiMo12O40@HKUST-1,PW12O40@HKUST-1和PMo6W6O40@HKUST-1,并利用X射线结构分析表征了化合物的结构,位于通道内的结晶H2O分子通过弱氢键作用与POMs阴离子连接,进一步稳定了该材料的结构[31]。
2009年,刘术侠等在上述Naruke课题组研究工作基础上利用已制备的Keggin型POMs(HnXM12O40,X=Si,Ge,P,As;M=W,Mo)为起始反应物,用(CH3)4NOH调节溶液pH值,在Cu(NO3)2·3H2O与H3BTC原位生成HKUST-1的同时将POMs成功封装到MOFs中,得到一系列POMs@HKUST-1杂多化合物,并用X射线单晶衍射表征了其结构(图2),命名为 NENU-n 系列(n=1~6;NENU=Northeast Normal University),其中在MOFs材料B孔中存在的结晶水分子及抗衡阳离子(CH3)4N+分别通过氢键和静电作用使POMs阴离子与 MOFs的结合更加牢固[32]。2011 年, 该课题 组又 以 CuCl2·2H2O,Na3PW12O40·12H2O为起始原料通过一步水热反应将 [PW12O40]3-封装到方钠石型MOFs骨架中制得POMs@MOFs型杂 多 化 合 物 H3[(Cu4Cl)3(BTC)8]2[PW12O40]3·(C4H12N)6·3H2O(NENU-11)。在该结构中,每个[PW12O40]3-与周围的6个(CH3)4N+通过静电作用结合[33]。Naruke和刘术侠等的研究代表了原位合成法制备POMs@MOFs(COFs)材料的2种典型的合成方法,即:(1)POMs、MOFs同时原位合成封装;(2)预先制备好POMs,然后将其封装到原位合成的MOFs中。

图2 在NENU-n(n=1~6)系列化合物中沿{001}面存在的2种介孔[32]Fig.2 Two types of mesopores in the compounds of NENU-n(n=1~6)series along the{001}sheet[32]
2006年,孟庆金课题组利用原位合成方法在乙腈/水混合溶剂中将质子化的大水簇H+(H2O)27和Keggin结构的多阴离子 [PW12O40]3-装载到由Co(Ⅱ)和4,4′-二吡啶-N,N′-二氧二水合物(dpdo)构筑的三维MOFs空腔中。X射线衍射结合理论计算结果表明,有大量的质子存在于质子化水簇的中心。每个水簇的周围有12个乙腈分子通过O-H…N键与水簇连接,在水簇和MOFs之间起到支撑的作用,另外,乙腈分子在水簇和 [PW12O40]3-之间也起到重要的连接作用,稳定了化合物的整体结构[34]。后续研究中,段春迎等又合成了一系列该体系的化合物,并确定了其单晶结构[35-36]。2008年,王秀丽课题组将CuCl2·2H2O 的乙醇溶液与 bpp(1,3-双(吡啶基)丙烷)的醋酸溶液混合搅拌,然后加入溶有POMs的DMF溶液,将[PM12O40]3-(M=W,Mo)一步封装到MOFs骨架中,并确定了其单晶结构[37]。后续工作中,该课题组又制备出一系列具有POMs@MOFs结构的化合物[38]。
2009年,本课题组也在水热条件下一步合成了系列3D阳离子框架[Ln(H2O)4(pydc)]44+(Ln=La,Ce和Nd;H2pydc=吡啶-2,6-二甲酸)封装 Keggin 型多阴离子[XM12O40]4-(X=Si,Ge;M=Mo,W)的化合物(图 3),并通过X射线单晶衍射表征了其结构。有意思的是[Ln(H2O)4(pydc)]44+利用配位的H2O通过O-H…O键与[XM12O40]4-结合进一步稳定了化合物的结构[39]。

图3 由[XM12O40]4-占据的Z字形排列的交叉通道的空腔[39]Fig.3 Cavities of intersected channels in a zigzag arrangement cccupied by[XM12O40]4-[39]
2010年,Gascon课题组在用水热法原位合成MIL-101(Cr)的混合溶液中加入H3PW12O40,并成功将其封装进MIL-101(Cr)介孔中制得双功能H3PW12O40@MIL-101(Cr)催化剂[40]。MIL-100(Fe)具有刚性沸石型晶体结构,此结构的2种介孔笼尺寸分别为2.5和2.9 nm,开孔分别为0.55和0.86 nm。2011年,Canioni课题组采用一步水热法成功将H3PMo12O40封装到MIL-100(Fe)的介孔笼中,制备得到H3PMo12O40@MIL-100(Fe)催化剂。POMs负载量为65%的H3PMo12O40@MIL-100(Fe)催化剂,其BET比表面积从MIL-100(Fe)的 2 800 m2·g-1降到 1 000 m2·g-1,孔容从 0.882 cm3·g-1降到 0.373 cm3·g-1, 气体分子仍然可以较容易的进入其孔笼中。重要的是,将该材料分别在水中和有机相中长时间放置,均没有造成客体分子H3PMo12O40的流失,这说明制备的H3PMo12O40@MIL-100(Fe)材料非常稳定[41]。
2011年,Hill课题组利用水热反应将具有合适尺寸的Keggin型多酸[CuPW11O39]5-一步封装到原位合成的HKUST-1的主体框架中,并得到了单晶结构,制得了与NENU同系列的客体分子为取代型Keggin杂多酸的POMs@MOFs材料[42]。对化合物晶胞的分析表明,MOFs中的大孔和小孔分别被POMs阴离子及其抗衡阳离子占据,其中有大约50%的大孔是空的。
2012年,Juan-Alcañiz课题组通过改变POMs的添加量,利用传统和微波加热的方法,将H3PW12O40一步封装到窗口尺寸较小的MIL-100(Cr)中,得到不同POMs负载量的H3PW12O40@MIL-100(Cr)材料,且在水/2-戊醇两相溶液体系中用微波加热的方法能使H3PW12O40的装载量达到最大(质量分数约为30%)。研究发现,微波加热与传统加热方法相比反应时间能从4 d缩减到3 h,而两相溶液体系既能保持H3PW12O40的完整性,又不至于影响到MIL-100(Cr)的有效合成[43]。
2012年,吴传德课题组先利用H3PW12O40与MnⅢCl-TPyP(TPyP=四吡啶基卟啉)在DMF溶剂中制备得到两性离子化合物{[MnⅢ(DMF)2TPyP](PW12O40)}2-,然后再与Cd(NO3)2·4H2O在DMF和醋酸的混合溶液中反应制备得到POMs封装型[PW12O40]@Cd-MnⅢ-TPyP层状框架材料,并确定了其单晶结构(图4)。化合物孔道中存在的DMF和H2O分子以及抗衡阳离子等通过氢键作用和静电作用对化合物结构的稳定起了重要的作用[44]。段春迎课题组在溶剂热反应条件下,通过一步反应将[BW12O40]5-装载到由Ni和手性配体L-或D-吡咯烷基-2-咪唑(PYI)原位反应构筑的手性MOFs骨架中,制备得到具有手性的POMs@MOFs型材料,并拿到了化合物的单晶结构。质子化的吡咯烷N原子与[BW12O40]5-形成的N-H…O氢键和抗衡阳离子与多阴离子之间的静电作用一起稳定了化合物的结构[45]。

图4 [PW12O40]@Cd-Mn-Porphyrin的结构[44]Fig.4 Structure of[PW12O40]@Cd-Mn-Porphyrin[44]
如图5所示,2012年,王恩波课题组以1-乙基-3-甲基咪唑溴化物([Emim]Br)为溶剂,首次利用离子热法将[Mo8O26]4-多阴离子一步封装到由Cu2+和1,4-二(1,2,4-三唑-1-甲基)苯(BBTZ)构筑的多孔 MOFs材料中,制备得到3例 [Mo8O26]@MOFs多酸基框架材料(TBA)2[Cu(BBTZ)2(x-Mo8O26)](x=β(1),α(2,3);TBA=四丁基氨阳离子)。离子热合成法为制备POMs@MOFs型材料提供了环保且有效的替代方案,研究认为大的模板剂利于大孔径结晶材料的制备[46]。2015年,该课题组利用水热反应将[HPMo12O40]2-多阴离子封装到由 Co2+和 4,4′-二(1,2,4-三唑-1-甲基)联苯(BBPTZ)构筑的MOFs材料中,制备得到 1例[HPMo12O40]@MOFs多酸基框架材料。结构研究发现其具有3D CdSO4(cds)型框架结构,且有2种不同类型的孔道互联。该材料被进一步用作石油氧化脱硫的催化剂,表现出较好的尺寸选择性和结构稳定性[47]。
图5 [Mo8O26]@MOFs材料的结构和通道[46]Fig.5 Structures and channels of[Mo8O26]@MOFs materials[46]
2016年,李阳光课题组利用水热法制得2例POMs@MOFs型化合物[Zn(bimbp)2]3[PMo12O40]2和[Bu4N][Zn3(bimb)4Cl(MoO4)][PMo12O40](化合物结晶水分子已省略;bimbp=4,4′-二(咪唑基)联苯;bimb=1,4-二(1-咪唑基)苯),并经过煅烧后制备得到有氮掺杂的石墨烯外壳覆盖的碳化钼新型纳米颗粒(MoCx@C-1)和无覆盖的纳米颗粒(MoCx-2)[48]。
近几年来,将POMs@HKUST材料进一步与分子筛、碳材料以及纤维材料等复合的研究也受到重视。2014年,高爽课题组利用水热反应将H6PMo9V3O40封装到原位制备的HKUST-1(MOF-199)骨架中,并与反应体系中存在的SBA-15介孔分子筛结合制得POMs@MOFs@SBA-15杂化材料[49]。赵建设课题组也用类似的方法合成得到MH3PMo6W6O40@MOF-199@MCM-41材料(M=Cr,Mn,Fe,Co,Ni,Cu,Zn,Cd)[50]。兰亚乾课题组则将H5PMo10V2O40@HKUST-1与石墨烯复合制备的材料用于修饰锂离子电池的阳极,对于Li和Na存储的先进电极材料的设计具有指导意义[51]。Obendorf课题组将[CuPW11O39]5-封装到原位生成的HKUST-1中,并在室温下将该材料成功通过醚化官能化自组装到纤维素基质(棉花)上,制得POMs-MOFs-cotton材料。经过计算,POMs在MOFs笼中的占有率约为13%[52]。
2015年,张富民等在常压、低温和无HF存在的条件下,成功将H3PW12O40作为客体分子封装进原位制备的MIL-100(Fe)的介孔笼中,得到H3PW12O40@MIL-100(Fe)型材料[53]。同年,刘术侠课题组在前期工作的基础上与本课题组合作,通过改变H3PW12O40的负载量和反应温度,并加入模板剂来调控NENU-3同系列的POMs@MOFs材料的晶面结构,以增加POMs在某一优势晶面的暴露,分别得到了四方体和八面体晶体(POMs在{100}的暴露远多于{111}晶面)(NENU-3a)(图6)。与前期合成的NENU-3不同,由于合成过程中没添加(CH3)4NOH,所以产物中抗衡阳离子只有H+而没有(CH3)4N+[54]。

图6 [PW12O40]@HKUST的组装及不同晶型展示出的优势晶面[54]Fig.6 Assembly of[PW12O40]@HKUSTand the dominant crystal facet showed by different crystals[54]
如图7所示,2015年林文斌课题组利用一种简单有效的电荷辅助自组装方法将Wells-Dawson型多阴离子[P2W18O62]6-封装进由[Ru(bpy)3]2+衍生化的二酸配体与Zr6(μ3-O)4(μ3-OH)4构筑的MOFs孔道中,并通过调控POMs的添加量制得一系列POMs@UiO材料[55]。

图7 封装型[P2W18O62]@UiO的合成示意图[55]Fig.7 Schematic representation for the synthesis of encapsulated[P2W18O62]@UiO[55]
2015年,Dolbecq课题组首次利用溶剂热法将Keggin 型 [PW12O40]3-,[PW11O39]3-和 Wells-Dawson 型[P2W18O62]6-多阴离子一步封装到原位法制备的UiO-67孔道中制得一系列POMs@UiO-67材料[56]。李丹课题组在Dolbecq等工作的基础上用原位合成方法制备得到PW12@UiO-67材料[57]。同年,王恩波课题组将过渡金属钴充当杂原子的[CoW12O40]6-簇装载到原位制备的MIL-101(Cr)空腔中,制得CoW12@MIL-101(Cr),并将其加到去离子水中超声处理,然后将该溶液与溶有钛酸异丙酯的正丁醇溶液混合,搅拌、过滤、干燥,最后将所得固体煅烧制得CoW12·Cr2O3@TiO2,该材料能很好地改善量子点太阳能电池的性能[58]。
2016年,Fazaeli课题组通过一步水热法将H3PW12O40装载到原位合成的ZIF-8腔体中,制备得到POMs@ZIF-8材料[59]。同年,卢灿忠课题组以CuCl和 1,2,4,5-四(4-吡啶基)苯(TPB)为起始原料,利用溶剂热方法,以H3PW12O40作为模板原位合成得到Cu-MOF装载POMs的材料。其中[PW12O40]3-作为客体占据沿c轴的较大通道,而沿{110}方向较窄的一维通道用于容纳溶剂分子和(CH3)2NH2+抗衡阳离子,其中(CH3)2NH2+与POMs阴离子的静电作用进一步稳定了化合物的结构[60]。
2016年,林文斌课题组将含4核Ni的多金属氧酸盐[Ni4(H2O)2(PW9O34)2]10-(Ni4P2)封装到由[Ir(ppy)2(bpy)]+和[Ru(bpy)3]2+衍生的二羧酸配体与ZrCl4原位合成法制备的一系列高稳定性、多孔UiO-MOF中,得到Ni4P2@MOF-1和Ni4P2@MOF-2,X射线粉末衍射表明MOF-1和MOF-2都具有大的四面体和八面体空腔,足以容纳尺寸较大的[Ni4(H2O)2(PW9O34)2]10-。在MOF-1和MOF-2每个分子式的空腔中存在6个或12个CF3CO2-以平衡电荷并通过静电作用稳定化合物结构[61]。
2016年, 闫鹏飞等以 CuCl2·2H2O,5-四唑间苯二甲酸(H3TZI)和 H3PM12O40·H2O(M=Mo,Si,W)为起始原料,在DMF/水的混合溶剂中通过溶剂热反应制得 POMs@rht-MOF-1系列封装型材料(HLJU1~3),并得到明确的单晶结构,首次证明Keggin型杂多酸可以装载到具有4种笼(A~D笼)的高度有序和多孔结构的rht-MOF-1的β-笼中(图8中的C笼)[62]。
近年来,COFs装载POMs的研究也受到多酸化学家的重视。2017年,何宏艳等将{(PW9)2M7}(M=Co,Ni)多阴离子原位装载到TTF(热固化三嗪基骨架)类COFs骨架中制得 POMs@TTF-F-COFs材料(图 9),并进一步研究了其催化性能[63]。

图8 HLJU1~3中4种金属有机笼示意图[62]Fig.8 Schematic representation for the four types of cages in HLJU1~3[62]

图9 POMs@TTF-F合成路线示意图[63]Fig.9 Schematic representation for the synthetic route of POMs@TTF-F[63]
2018年,Nadeem课题组将含有混价Co(Ⅲ)和Co(Ⅱ)离子的Keggin型多阴离子[CoⅡCoⅢW11O39(H2O)]7-(Co2)和[Co4(PW9O34)2(H2O)2]10-(Co4)用原位合成法封装到MIL-100(Fe)的介孔笼中,制得Co2@MIL-100(Fe)和 Co4@MIL-100(Fe)两种材料(图 10),POMs 的最大负载量能分别达到48%(Co2)和54%(Co4)[65]。

图10 POMs封装型MIL-100(Fe)的合成示意图[65]Fig.10 Schematic representation for the synthesis of encapsulated POMs in MIL-100(Fe)[65]
2018年,Das课题组将以过渡金属Co为中心杂原子的Keggin型多阴离子[CoW12O40]6-封装进原位合成法制备的ZIF-8空腔中,构筑了[CoW12O40]@ZIF-8主-客体材料[66]。朱广山课题组则在ZnO基板上使用含有H5PMo10V2O40的超薄ZIF-8薄膜用原位合成方法制备了不同POMs负载量的核-壳材料POM@ZIF-8@ZnO[67]。Yadav课题组也利用一步法将H3PW12O40装载到原位合成法制备的ZIF-8空腔中,制备了不同POMs负载量的封装型材料[68]。
2018年,王瑞虎课题组利用简便的原位自组装策略,将H3PMo12O40装载到ZIF-67中,该H3PMo12O40@ZIF-67材料在空气中煅烧可得到由超细微晶组成的Co3O4/CoMoO4多孔纳米笼,为表面氧化还原反应提供丰富的活性位点[69]。2018年,肖玲课题组也利用溶剂热的合成方法将K6P2W18O62装载到原位合成的UiO-66介孔中,通过调控调节剂三氯乙酸(TCA)的添加量,制备得到一系列POMs@UiO-66材料,并将其应用于阳离子型染料的选择性吸附[70]。
2019年,谢文磊等首次将Keggin型杂多酸H3PW12O40,H4SiW12O40和 H3PMo12O40装载到原位合成方法制备的UiO-66-2COOH金属有机框架中,并通过阴离子交换法进一步与磺酸类离子液体结合,制备得到AILs/POM/UiO-66-2COOH(AILs=acidic ionic liquids)材料(图 11)[71]。
2019年,刘春森等首次制备出能同时产氢和产氧的非贵金属可见光驱动分解水催化剂[72]。该催化剂是利用原位合成方法将Keggin型的 [W12O40]8-和Lindqvist型的 [W6O19]2-多阴离子嵌入阳离子型Cu-MOFs孔道中,通过静电作用制备得到基于{CuI24(μ3-Cl)8(μ4-Cl)6}的双功能化POMs@MOFs材料(ZZUIL-1)(图 12)。
2019年,刘术侠课题组以NaVO3、乙酸镍和4,4′-联吡啶(4,4′-bpy)为起始原料,在乙醇/水混合溶剂中利用冰醋酸调控溶液的pH值,首次将具有混合价态的{V16}多钒氧簇装载到原位合成的[Ni(4,4′-bpy)2]构筑的 MOFs中得到[VⅣ7VⅤ9Cl]@MOFs型超薄纳米片(约 5 nm)(NENU-MV-1a)[73]。

图11 AILs/POM/UiO-66-2COOH的合成示意图[71]Fig.11 Schematic representation for the synthesis of AILs/POM/UiO-66-2COOH[71]

图12 Keggin/Lindqvist双官能化POMs@MOFs的合成及光驱动催化产氢产氧示意图[72]Fig.12 Schematic representation for the synthesis of dual-functionalized mixed Keggin/Lindqvist type POMs@MOFs and as photocatalysts for both H2 and O2 evolution[72]
从上述原位合成法制备POMs@MOFs(COFs)材料的研究可以看出,作为客体的POMs主要是稳定性较高的Keggin型多酸,也有少量的Wells-Dawson型和Lindqvist型多酸,这主要与原位合成MOFs所需的苛刻条件有关,而用到的MOFs主体材料窗口和空腔相对都较小,不易用浸渍法和机械研磨法制备POMs封装型材料。
2.2 浸渍合成法
浸渍合成法是预先将要负载的POMs溶解于去离子水或有机溶剂中,制得多酸溶液,然后加入一定量的MOFs(COFs)载体,在一定温度下搅拌后静置,通过MOFs(COFs)的静电吸附和分子间的扩散作用将POMs装载进MOFs(COFs)的孔道中。这种方法的优点是避免了原位合成法中MOFs(COFs)合成所需的严苛条件对POMs结构的破坏,且制备的POMs@MOFs(COFs)材料中POMs分散比较均匀,缺点是需要根据POMs的尺寸选取具有合适窗口和孔道结构的MOFs(COFs)材料,MOFs(COFs)的窗口过小或POMs尺寸过大,POMs则无法进入MOFs(COFs)的腔体内部,而如果MOFs(COFs)窗口过大,POMs则容易流失,很难在其中稳定分散[74-86]。
2005年,Férey课题组在Science上首次报道了具有2种不同介孔笼结构的MIL-101(Cr),2种笼内孔径分别为2.9 nm(窗口为五边形1.2 nm×1.2 nm)和3.4 nm(窗口为六边形1.6 nm×1.5 nm)。 MIL-101(Cr)独特的分子结构、高的比表面积,使其成为封装POMs的理想载体,并首次将Keggin型多酸K7PW11O40·n H2O(直径约 1.3 nm)利用浸渍法装载到MIL-101(Cr)的大孔中,成功阻止了其进入小孔中,这为制备封装型POMs@MOFs非均相催化剂开辟了新途径。抗衡阳离子K+和H+与POMs阴离子处于MIL-101(Cr)相同的孔腔中,并通过静电作用使化合物结构得到稳定[74]。
2008年,Kholdeeva课题组首先把过渡金属取代的 M-POMs(NaH4PW11TiO40或 TBA4HPW11CoO39)溶解到乙腈溶液中,然后将水热条件下制备的经典MOFs材料MIL-101(Cr)加入到上述溶液中,搅拌后室温下静置,最后经过滤、洗涤、干燥,得到POMs@MIL-101(Cr)材料[75]。
2013年,Balula课题组将含镧系金属元素的多酸[Ln(PW11O39)2]11-(Ln=Eu3+和Sm3+)溶解到水溶液中,然后将其加入到预先制备好的MIL-101(Cr)中,室温下搅拌24h,过滤、洗涤、干燥得到浸渍法制备的[Ln(PW11O39)2]@MIL-101(Cr)材料[76]。同年,Freire 课题组利用浸渍法将预先制备的MIL-101(Cr)加入到TBA-[PMo10V2O40]的乙腈溶液中室温下搅拌4 d,经过滤、洗涤、干燥后制得PMo10V2@MIL-101(Cr)材料,并进一步研究了其电催化性能[77]。2014年,该课题组又将缺位的Keggin型多酸[A-PW9O34]9-(PW9)利用浸渍法装载到MIL-101(Cr)中制得PW9@MIL-101(Cr),并将其用于催化石油氧化脱硫反应[78]。
2016年,李斌等设计了具有红光发射的带有正电荷的溴化乙锭(EB)有机单体,首次制备出带正电荷的有机二维多孔晶体骨架材料EB-COF∶Br,然后利用阴离子交换法将PW12O403-装载到COFs中制备得到EB-COF∶PW12材料(图 13)。 EB-COF 与 PW12O403-之间通过静电作用稳定了POMs@COFs材料的结构[79]。

图13 阴离子交换法制备[PW12O40]@EB-COF的示意图[79]Fig.13 Schematic representation for the synthesis of[PW12O40]@EB-COF by anionic exchange[79]
2017年,刘术侠课题组将预先合成的MIL-101(Cr)加入到溶有H3PW12O40的水溶液中,室温下搅拌2 d后,经分离、洗涤、干燥制得POMs@MIL-101(Cr)材料,并对MIL-101(Cr)装载POMs的能力进行了研究,将H3PW12O40分别装入MIL-101(Cr)的大笼和小笼中,并推测每个大笼和小笼中可分别装载5个和3 个 H3PW12O40[80]。
2017年,Farha课题组通过浸渍法将Keggin型杂多酸H3PW12O40引入到Zr基MOFs材料NU-1000的介孔中,制得PW12@NU-1000杂化材料[81]。2018年,该课题组在上述工作基础上,进一步合成了经超临界CO2活化处理的PW12@NU-1000-scCO2和直接高温处理的PW12@NU-1000-120℃材料[82]。
2018年,付宏刚课题组首先合成得到ZIF-67,通过在甲醇溶液中长时间搅拌加热的方法使ZIF-67部分解离形成缺陷,将较小的窗口打开,然后成功将[PMo12O40]3-装载到ZIF-67的介孔笼中,这种方法为POMs和MOFs的结合提供了一种新的合成策略。该[PMo12O40]@ZIF-67材料经过煅烧得到的小尺寸的双金属羰化物MoxCoxC[83]。Monakhov课题组也于同一年报道了PMo12O40@ZIF-67材料的合成方法[84]。
2019年,Shul课题组将不同量的Keggin型多酸H3PW12O40分别溶解到去离子水中,然后将预先合成的定量ZIF-8纳米颗粒加入到上述溶液中超声处理后,在室温下搅拌,离心分离,洗涤、干燥,得到具有核-壳结构的酸碱双功能非均相催化剂H3PW12O40@ZIF-8[85]。
如图14所示,2019年,朱广山课题组利用前期合成的具有咪唑鎓官能团的离子多孔芳香骨架材料iPAF-1(iPAF=ionic porous aromatic framework),通 过浸渍法将系列PMoV杂多酸(H3PMo12O40,H4PMo11VO40,H5PMo10V2O40,H6PMo9V3O40和(NH4)5H6PMo4V8O40)封装进iPAF-1孔道中,制备得到[PMoV]@iPAF-1材料[86]。
从上述浸渍法制备POMs@MOFs(COFs)材料的报道可以看出,作为客体的POMs同样以饱和的Keggin型多酸为主,也有少量的缺位型多酸 (例如[A-PW9O34]9-),这与浸渍法所需的条件比较温和有关,而装载主体MOFs(COFs)材料的窗口和空腔相对较大 (例如 MIL-101,NU-1000 和 EB-COF,iPAF-1),POMs比较容易进入MOFs(COFs)空腔,也可以用适当方法打开较小的窗口,然后再装载POMs(例如ZIF-67)。

图14 iPAF-1的合成示意图[86]Fig.14 Schematic representation for the synthesis of iPAF-1[86]
2.3 机械研磨合成法
机械研磨合成法是指在无溶剂(纯固相)或者微量溶剂辅助条件下,将机械研磨的机械能转化为化学能使其迅速发生固相化学反应的一种合成方法,具有绿色、节能、简单、高效的优点,缺点是该方法要求反应的活化能不能太高,产物的收率和结晶度也不是很理想,并且很难从微观分子水平去解释合成反应机理[87-91]。
2013年,刘术侠课题组利用不同的方法合成了[PMo10V2O40]@HKUST材料 (NENU-9和NENU-9N)。其中利用经典的水热原位合成方法可以得到体积较大,适合X射线单晶衍射测试的单晶样品。而利用溶液搅拌法,通过调控铜盐的类型 (硝酸盐和醋酸盐)以及反应溶液的pH值可以制得直径分别为1 300、550和80 nm的微晶材料(NENU-9N)。此外,研究者还探索了更简便的机械研磨合成方法,将Cu(NO3)2·3H2O和H5PMo10V2O40溶解到水溶液中并调节pH值到2~4。然后将溶剂挥发并与H3BTC一起研磨大约5min,经洗涤、干燥后制得直径大约为150 nm的POMs@MOFs微晶样品。这为该类材料的便捷、快速合成提供了一条有效的途径[87]。
如图15所示,2014年,本课题组和王博教授合作利用简单的机械球磨法,以ZnO,2-甲基咪唑和Keggin型POMs为起始原料,通过改变POMs的类型和调控其添加量(0.2~1.6 mmol),率先实现了Keggin结构POMs在ZIF-8腔体中的装载,合成了H3PW12O40@ZIF-8(BIT-1),H4SiW12O40@ZIF-8(BIT-2)和H3PMo12O40@ZIF-8(BIT-3)(BIT=Beijing Institute of Technology)装载型材料[88]。

图15 封装型BIT-n@ZIF-8的固相合成示意图[88]Fig.15 Schematic representation for the mechanochemical synthesis of encapsulated BIT-n@ZIF-8[88]
如图16所示,本课题组于2017年以ZnO,(NH4)2SO4,2-乙基咪唑,4,5-二氯咪唑和 Keggin 型钒取 代 磷 钼 酸 PMoV(H4PMo11VO40,H5PMo10V2O40和H6PMo9V3O40)为起始原料,通过机械研磨将POMs装载到具有rho结构的ZIF孔道中,制备得到PMoV@rho-ZIF 型材料[89]。

图16 封装型PMoV@rho-ZIF的固相合成示意图[89]Fig.16 Schematic representation for the mechanochemical synthesis of encapsulated PMoV@rho-ZIF[89]
2018年,刘术侠课题组将Cu(NO3)2·3H2O和H3PW12O40溶解到去离子水中并调节溶液pH值到4,挥发干燥后与H3BTC混合研磨大约5 min,经洗涤、干燥后制得直径大约为190 nm的POMs@MOFs微晶样品,收率约91%。随后,研究者探索了合成方法的改进,在反应混合物中加入少量乙醇做辅助溶剂研磨5 min后,制得直径为85~130 nm的POMs@MOFs微晶样品,且收率提高到95%[90]。
2019年,王尧宇课题组探索了新颖的研磨和原位热压相结合的合成方法,将H3PW12O40(PW12)在短时间内迅速装载到 In3+和 3,3′,5,5′-联苯四甲酸(H4BPTC)形成的新型MOFs材料MFM-300(In)中,制备得到PW12@MFM-300(In)材料。这种无溶剂制备工艺既环保又节约能源和时间,符合绿色化学合成的要求[91]。
从上述机械研磨合成法制备POMs@MOFs(COFs)材料的文献报道可以看出,作为客体的POMs都是饱和的Keggin型多酸,而作为负载主体的MOFs合成相对容易,需要的活化能不高,这主要受机械研磨法所提供的能量限制。
3 POMs@MOFs(COFs)材料的性能
POMs@MOFs(COFs)材料既具有优美的拓扑结构又具有POMs和MOFs丰富的双功能特性,还具有开放式孔道的特点及较大的比表面积和较高的分散度。POMs@MOFs(COFs)材料的这些特性大大拓展了该类材料的应用范围,包括光电催化、传统有机催化、染料吸附、药物缓释、质子传导、气体传感以及电极材料制备等众多领域 (图17)。下面对POMs@MOFs(COFs)材料的主要性能及应用分类总结,并作简要介绍。

图17 POMs@MOFs(COFs)材料的性能示意图Fig.17 Schematic representation for the performance of POMs@MOFs(COFs)materials
3.1 光电催化性能
2008年,王秀丽课题组合成的POMs@MOFs型材料在环境温度下表现出光致发光性质,并可用于修饰碳糊电极对亚硝酸盐的还原,表现出良好的电催化活性[37]。2016年,卢灿忠课题组利用合成的Cu-MOFs装载[PW12O40]3-的材料做催化剂,在H2O2存在下,用300 W的Xe灯作为紫外-可见光源照射,发现该材料能高效吸附并降解甲基橙(MO)染料[60]。
2012年,王恩波课题组研究了多酸基框架材料(TBA)2[Cu(BBTZ)2(α-Mo8O26)]在紫外光激发下的光催化产氢性能,平均产氢速率为0.78μmol·h-1,空白实验以及单独使用(TBA)4(Mo8O26)为催化剂则没有氢气生成,进一步证明合成的POMs@MOFs材料对光驱动催化产氢具有活性[46]。
Freir课题组研究了[PMo10V2]@MIL-101(Cr)封装型材料的电催化性能。结果证明,在电催化氧化抗坏血酸的反应中,该材料修饰的电极相较于游离的H5PMo10V2O40修饰的电极催化性能有显著提升(图18)。[PMo10V2]@MIL-101(Cr)修饰后的电极材料还能同时检测抗坏血酸和多巴胺[77]。

图18 PMo10V2@MIL-101(Cr)结构及电催化性能[77]Fig.18 Structure and electrocatalytic property of PMo10V2@MIL-101(Cr)[77]
2015年,林文斌课题组利用Wells-Dawson型多阴离子[P2W18O62]6-与UiO-MOFs形成的材料用作光解水催化剂。该材料是首例由可见光驱动的分解水产氢POMs@MOFs型催化剂[55]。2017年,何宏艳等将合成的{(PW9)2M7}@TTF-F-COFs(M=Co,Ni)材料用作氧气的电还原反应(ORR)催化剂,在中性介质中显示出可比拟Pt/C电极的催化活性以及比Pt/C电极更高的稳定性[63]。
2018年,张志明等将Wells-Dawson型多阴离子[P2W15V3O62]9-,[P2W17(NiOH2)O61]8-和[P2W17(CoOH2)O61]8-与MIL-101(Cr)制备的封装型材料用于催化光驱动分解水反应,产氢效率最高可达25 578μmol·h-1·g-1[64]。 同一年,Nadeem 课题组将[CoⅡCoⅢW11O39(H2O)]@MIL-100(Fe)和[Co4(PW9O34)2(H2O)2]@MIL-100(Fe)材料用作水氧化催化剂,在可见光照射下与单纯的POMs催化剂相比,具有更好的光驱动催化产氧性能[65]。
如图19所示,2018年,Das课题组将构筑的[CoW12O40]@ZIF-8用作高效电分解水氧化催化剂,实现了饱和型POMs的电催化产氧。该催化剂在中性pH 条件下的转化频率(TOF)为 10.8 molO2·molCo-1·s-1,具有高效的电催化水氧化活性,且经1 000次循环使用仍无明显活性降低[66]。
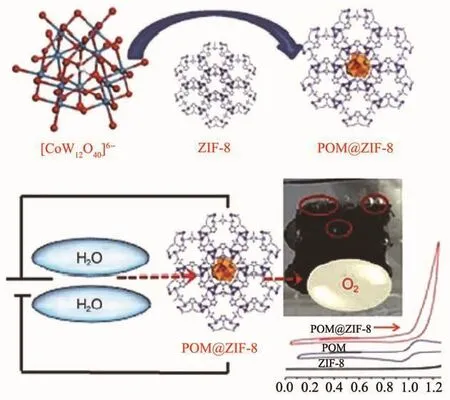
图19 封装型[CoW12O40]@ZIF-8的合成及电催化水氧化示意图[66]Fig.19 Schematic representation for the synthesis of encapsulated[CoW12O40]@ZIF-8 and electrocatalytic water oxidation[66]
最近,将主客体POMs@MOFs材料作为前驱体制备电解水纳米催化剂的研究也取得了较大的进展。2016年,李阳光课题组将制备的氮掺杂石墨烯外壳覆盖的碳化钼纳米颗粒MoCx@C-1用作电驱动水分解产氢的催化剂。实验证明,在酸性介质中MoCx@C-1显示出可比拟20%Pt/C商业化产品的产氢催化活性,且具有非常出色的长期循环稳定性。而在10 mA·cm-1电流密度下表现出的79 mV的过电位,使其成为目前在酸性介质中最好的基于MoCx的电驱动产氢催化剂之一,其性能要优于无覆盖的纳米颗粒MoCx-2[48]。2018年,王瑞虎课题组将由H3PMo12O40@ZIF-67材料煅烧得到的Co3O4/CoMoO4多孔纳米笼用作催化剂,在碱性条件下的电分解水氧化活性得到了显著的提高,其性能可与商品化的贵金属IrO2相媲美,是多酸基MOFs材料衍生出的优异电分解水氧化催化剂[69]。2018年,付宏刚课题组将[PMo12O40]@ZIF-67材料煅烧制备了双金属羰化物MoxCoxC,具有良好的电催化水分解产氢、产氧性能[83]。以POMs@MOFs材料为前驱体制备的纳米催化剂催化效果较好,但同时也存在原子经济性差的缺点,不符合绿色合成的理念,合成方法有待改进。
3.2 传统有机催化性能
POMs@MOFs(COFs)材料在传统有机催化反应领域主要体现在酸碱催化性能和氧化催化性能。下面分别从这2个方面来总结、介绍封装型POMs@MOFs(COFs)材料的催化性能。
3.2.1 酸碱催化性能
2006年,孟庆金课题组合成了封装有质子化大水簇 H+(H2O)27和 [PW12O40]3-的 Co-dpdo三维 MOFs[34]。在后续的工作中,段春迎等对该体系的材料性能进行了深入研究,并将其作为非均相酸催化剂用于磷酸二酯水解反应[35-36]。

图20 NENU-3a催化酯水解的结果[32]Fig.20 Results for the hydrolysis of ester catalyzed by NENU-3a[32]
2009年,刘术侠等合成了系列POMs@HKUST-1 杂多化合物(POMs=HnXM12O40,X=Si,Ge,P,As;M=W,Mo)。如图 20所示,H3PW12O40作为 Keggin系列多酸中最强的Brønsted酸,装载到HKUST-1中形成的H3PW12O40@HKUST-1(NENU-3a)在水溶液中对对乙酸甲酯和乙酸乙酯的水解反应表现出良好的尺寸选择性和酸催化活性,转化率明显高于苯甲酸甲酯,苯甲酸乙酯和丙酸-4-甲基苯酯[32]。后续工作中,该课题组 又 合 成 了 H3[(Cu4Cl)3(BTC)8]2[PW12O40]3·(C4H12N)6·3H2O(NENU-11),NENU-11对神经毒气类似物甲基膦酸二甲酯(DMMP)表现出良好的吸附行为。得益于客体分子H3PW12O40的强Brønsted酸性和MOFs材料Lewis酸性的协同催化活性,DMMP可以通过水解反应顺利分解降解,且经过10轮循环以后催化剂结构仍能保持稳定[33]。
2010年,Gascon课题组考察了H3PW12O40@MIL-101(Cr)材料的酸催化性能,将其用于苯甲醛和氰乙酸乙酯的缩合反应、乙酸和正丁醇的酯化反应和甲醇脱水成醚的反应[40]。2015年,Obendorf课题组将[CuPW11O39]@HKUST-1材料,通过醚化官能化自组装到纤维素基质(棉花)上,制备得到POMs-MOFscotton材料。该材料能从己烷溶液中高效地除去甲基对硫磷(0.089 mg·g-1),而MOFs-cotton材料的去除率仅为(0.054 mg·g-1),研究者进一步讨论了反应机理,认为通过在MOFs材料中引入POMs,MOFs中需要更多暴露的Cu2+来补偿POMs多阴离子的电荷,增加了与甲基对硫磷结合的Lewis酸活性位点。POMs的 Brønsted酸性和 MOFs的 Lewis酸性协同作用增强了[CuPW11O39]@HKUST-1材料的酸催化甲基对硫磷水解的性能,使去除效率显著提高[52]。
张富民等将合成的H3PW12O40@MIL-100(Fe)材料用于催化正己醇与乙酸的酯化反应,正己醇转化率高达96.3%,且催化剂经4次循环利用后活性没有明显降低,同时未发现活性组分H3PW12O40的溶脱现象,表明该催化剂具有良好的稳定性[53]。
如图21所示,2015年,刘术侠课题组通过调控NENU-3系列POMs@MOFs材料的晶面结构,得到了四方体和八面体结构的晶体,特定的晶体在催化制备生物柴油过程中具有增强的晶面催化活性位点和对反应底物的吸附性能,证明H3PW12O40在{100}晶面(四方体晶体)暴露的远多于{111}晶面(八面体晶体),大大提高了其Brønsted酸催化活性,解决了由于反应底物分子过大而无法与催化剂活性位点充分接触的问题,长链脂肪酸(C12~C22)在制备生物柴油过程中的转化率均超过90%[54]。

图21 NENU-3不同晶面催化酯化反应的结果[54]Fig.21 Esterification reactions catalyzed by different crystal facets of NENU-3[54]
2018年,Yadav课题组将 H3PW12O40@ZIF-8应用于苯甲酸酐与肉桂醇的酸催化酯化反应[68]。2019年,谢文磊等将AILs/POMs/UiO-66-2COOH材料(POMs=H3PW12O40,H4SiW12O40,H3PMo12O40) 用于酸性植物油的酯交换反应制备生物柴油[71]。2019年,Shul课题组将具有核-壳结构的酸碱双功能非均相催化剂H3PW12O40@ZIF-8作为环境友好型非均相催化剂,用于菜籽油与甲醇的酯交换,生产出高质量的生物柴油[85]。
3.2.2 氧化催化性能
2008年,Kholdeeva课题组合成了MIL-101(Cr)装载Keggin型多酸阴离子[PW11CoO39]5-和[PW11TiO40]5-的催化剂,并以O2及H2O2为氧化剂氧化α-蒎烯制备马鞭草烯酮。在温和反应条件下,当α-蒎烯转化率为20%时,马鞭草烯酮的选择性达80%。2种复合型催化剂在重复利用过程中,POMs均没有明显的溶出现象发生,且能维持高的活性和选择性[75]。
2011年,Hill课题组制备了[CuPW11O39]@MOF-199材料,并将其用于O2(空气)作氧化剂的硫醇和H2S选择性氧化反应 (图22)。在水相H2S氧化为S8的过程中,单独使用K5[CuPW11O39]或者MOF-199没有S8固体生成,而且[CuPW11O39]@MOF-199的稳定性比单纯的POMs和MOFs都要高。控制实验发现,[PW12O39]@MOF-199材料在该反应中表现出非常低的催化活性。以上实验结果表明,[CuPW11O39]5-中的Cu2+应该是该催化剂的反应活性中心。而在硫醇的氧化实验中,MOF-199中的Cu2+中心和封装的[CuPW11O39]5-多阴离子之间的静电作用在稳定材料的同时,使反应活性位点得到充分暴露,大大增强了[CuPW11O39]@MOF-199材料的催化活性[42]。

图22 [CuPW11O39]@HKUST-1的硫醇和H2S选择性氧化降解催化性能[42]Fig.22 Selective oxidative decontamination of thiols and H2Scatalyzed by[CuPW11O39]@HKUST-1[42]
2012年,吴传德课题组将 [PW12O40]@Cd-MnⅢ-TPyP层状框架材料用于清除染料,并进一步用作苄基烷烃氧化反应的催化剂。在以叔丁基过氧化氢(TBHP)为氧化剂条件下对乙基苯的选择性催化氧化中,苯乙酮作为唯一产物收率能达到92.7%。在该氧化反应中,单独使用H3PW12O40为催化剂时,没有氧化产物生成,而当使用MnⅢCl-TPyP作为均相催化剂时,苯乙酮的收率仅为73.6%。以上控制实验结果表明,乙基苯选择性催化氧化的催化活性中心为MnⅢ,而均相的MnⅢCl-TPyP催化剂容易形成惰性的μ-氧代金属卟啉二聚体而导致失活。封装型的[PW12O40]@Cd-MnⅢ-TPyP催化剂中活性中心MnⅢ被大的客体分子H3PW12O40隔离,所以在催化反应特别是循环实验中能保持其催化活性[44]。
如图23所示,段春迎课题组将[BW12O40]5-装载到由Ni和手性配体L-或D-吡咯烷基-2-咪唑(PYI)构筑的手性MOFs骨架中。该材料被用作芳基烯烃的不对称二羟基化反应的两亲性催化剂,显示出优异的立体选择性[45]。

图23 [BW12O40]@MOFs合成及催化性能示意图[45]Fig.23 Schematic representation for the synthesis and catalytic property of[BW12O40]@MOFs[45]
2013年,Balula课题组以Eu(PW11)2@MIL为催化剂,H2O2做氧化剂,乙腈为溶剂用于苯乙烯的氧化催化,反应3 h后,苯乙烯转化率87%,产物苯甲醛的选择性达到60%,经4 h后苯乙烯几乎完全转化。另外,他们还研究了微波辅助条件下的烯烃氧化反应,发现与传统加热的反应条件相比,在更短的反应时间内催化剂活性保持不变的情况下,产物醛的选择性都有所增加[76]。2014年,Freire课题组将MIL-101(Cr)装载缺位多阴离子[A-PW9O34]9-(PW9)的材料PW9@MIL-101(Cr)用于催化石油氧化脱硫反应[78]。高爽课题组将H6PMo9V3O40@HKUST与SBA-15介孔分子筛结合制得POMs@MOFs@SBA-15杂化材料。该材料在由氧气氧化苯制备苯酚的反应中表现出非常高的选择性和较高的转化数和苯酚收率[49]。
2015年,王恩波课题组将合成的[HPMo12O40]@MOFs多酸基框架材料用作石油氧化脱硫的催化剂,表现出较好的尺寸选择性和结构稳定性[47]。赵建设课题组也将合成的Metal-POM@MOF-199@MCM-41材料用作氧气氧化石油脱硫的催化剂[50]。
2016年,闫鹏飞等将具有明确单晶结构的H3PM12O40@rht-MOF-1(M=Mo,Si,W)系列封装型材料(HLJU1-3)用作吸附材料及催化剂。高度分散的多阴离子和多孔结构提高了罗丹明B(RhB)和结晶紫染料的吸收效率。该材料还被用作TBHP为氧化剂的苄基烷烃的选择性氧化为相应酮的反应,具有较高的转化率和选择性(图24)[62]。

图24 不同尺寸的烷基苯氧化为相应的酮[62]Fig.24 Oxidation reactions of alkylbenzenes with different sizes for the formation of corresponding ketones[62]
2016年,Fazaeli课题组将制备的 H3PW12O40@ZIF-8用于催化硫化物的选择性氧化,通过调控溶剂类型和氧化剂H2O2的用量,可以由硫醚催化氧化分别来制备亚砜和砜,而且该催化剂经3次循环利用活性无明显降低[59]。
如图25所示,2017年,Farha课题组将 PW12@NU-1000材料用作H2O2为氧化剂的2-氯乙基乙基硫醚(CEES,芥子气类似物)氧化降解反应的催化剂。单独使用H3PW12O40为均相催化剂时,反应时间很长(约90 min),但部分氧化产物2-氯乙基乙基亚砜(CEESO)为唯一产物,即使增加H2O2用量结果也是一样。而当单独使用NU-1000为催化剂时,产物则以过氧化的2-氯乙基乙基砜(CEESO2)为主,如果加大H2O2用量,则可以使CEESO2成为唯一的产物。当使用PW12@NU-1000作为非均相催化剂时,在很短的时间内CEES就会完全转化 (约 20 min),且CEESO是主要产物。以上实验结果充分证明了PW12@NU-1000材料中POMs和MOFs的协同催化作用[81]。

图25 PW12@NU-1000催化CEES氧化降解[81]Fig.25 Oxidative decontamination of CEEScatalyzed by PW12@NU-1000[81]
2018年,Farha课题组又进一步研究了PW12@NU-1000材料在不同的活化条件下,对其催化性能的影响。研究发现,经超临界CO2活化处理的PW12@NU-1000-scCO2与直接高温处理的PW12@NU-1000-120℃相比,在CEES的选择性氧化降解反应中表现出更高的催化活性和选择性[82]。
2018年,李丹课题组制备了PW12@UiO-67材料,并将其应用于石油深度氧化脱硫反应(ODS)的催化剂。在硫的含量为1 g·L-1的模拟石油中,二苯并噻吩的去除率在1 h内能到到99.5%,且催化剂经8次循环实验后反应活性没有明显降低(图26)[57]。

图26 PW12@UiO-67催化氧化脱硫反应示意图[57]Fig.26 Schematic representation of oxidative desulfurization using PW12@UiO-67 as catalyst[57]
本课题组于2017年将装载型PMoV@rho-ZIF(PMoV=H4PMo11VO40,H5PMo10V2O40和 H6PMo9V3O40)材料用于H2O2为氧化剂条件下高选择性催化氧化硫醚为相应亚砜反应的催化剂。结果表明,当单独使用PMoV为均相催化剂时,产物亚砜的选择性较低,而单独使用rho-ZIF为催化剂则催化活性很差。PMoV@rho-ZIF材料作为POMs和MOFs的协同非均相催化剂则能很好的解决以上问题,既保持催化剂的高活性又具有高的选择性。该催化剂至少能循环使用4次而没有明显的活性降低。该研究对于开发更具活性和选择性的多酸基MOFs纳米催化剂具有重要意义[89]。
2018年,刘术侠课题组将H3PW12O40@HKUST微晶材料(NENU-3N)用于催化苯酚的降解反应,在POMs和MOFs的协同催化作用下实现了高达97%的转化率和88%的矿化率。此外,对比实验表明,NENU-3N催化剂的尺寸减小能改善其催化性能,且催化剂具有较高稳定性,易于回收并循环利用5次而不会损失催化活性(图27)[90]。
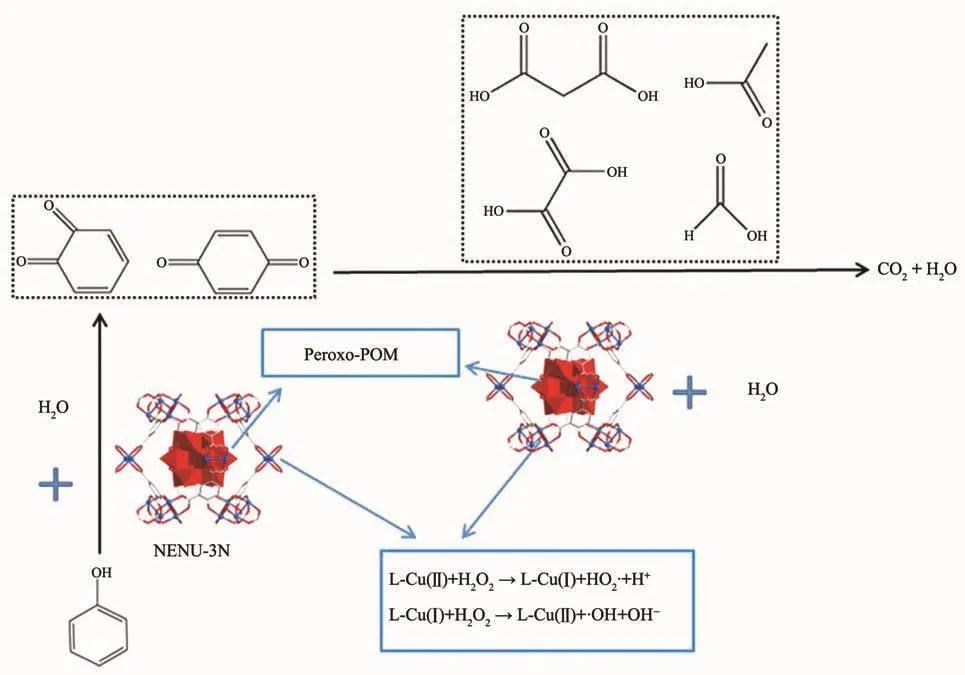
图27 苯酚催化降解反应示意图[90]Fig.27 Schematic representation of the catalytic degradation process of phenol[90]
2019年,刘术侠课题组将封装有混合价态的{V16}多钒氧簇 MOFs材料[VⅣ7VⅤ9Cl]@Ni-MOFs型超薄纳米片(约5 nm)(NENU-MV-1a)用于空气为氧化剂,异丁醛为牺牲剂的烯烃环氧化反应催化剂,表现出了比块状晶体(转化率35%)更优异的催化性能(转化率95%)。研究者还讨论了催化剂结构-性能的关系,并提出POMs和MOFs协同催化反应的机理。该研究对于开发更具活性和选择性的混合价金属-氧基团簇的MOFs纳米催化剂具有重要意义[73]。
如图28所示,2019年,朱广山课题组将合成的系列 [PMoV]@iPAF-1材料用作O2为氧化剂的氧化脱硫反应,其中(NH4)5H6PV8Mo4O40@iPAF-1在真实汽油和柴油染料的分子氧氧化脱硫方面具有很高的效率,在反应5 h后真实汽油中硫含量由996 mg·L-1降到 20.68 mg·L-1,柴油中的硫含量由 1 282 mg·L-1降到20.48 mg·L-1,且催化剂经7次循环利用后无明显活性降低[86]。

图28 POMs@iPAF-1催化剂组装示意图[86]Fig.28 Schematic representation of the assemble of POMs@iPAF-1 catalyst[86]
3.3 其它性能和应用
2014年,本课题组和王博教授合作制备的H3PW12O40@ZIF-8(BIT-1),H4SiW12O40@ZIF-8(BIT-2)和H3PMo12O40@ZIF-8(BIT-3)材料对亚甲基蓝有较好的吸附性能(饱和吸附量达到810 mg·g-1),而且具有对抗癌药物5-氟尿嘧啶(5-FU)较好的缓释性能(BIT-1缓释时间超过50 h,缓释率93%)(图29)。实验证明,该材料可同时作为优异的吸附剂和药物缓释剂[88]。
2017年,刘术侠课题组研究了H3PW12O40@MIL-101(Cr)材料的质子传导性能。结果表明,与Z形通路相比,线性通路能提供约2.1倍的质子扩散速率。用柔性多胺进一步改性的H3PW12O40@MIL-101(Cr)材料电导率能达到 1.52×10-2S·cm-1[77]。
2017年,李斌等将H3PW12O40装载到新合成的COFs材料中,制得H3PW12O40@EB-COFs材料。该材料的纳米孔结构及其表面不均匀的电场分布对质子和电子具有局部的限域作用。通过调节孔道内的电场分布,可以改变材料对质子的传导率。该H3PW12O40@EB-COFs材料在有机半导体器件和光电传感中具有潜在的应用价值[79]。

图29 不同材料对5-FU的缓释性能[88]Fig.29 5-FU controlled release by different materials[88]
2018年,肖玲课题组将合成的K6P2W18O62@UiO-66材料用于阳离子型染料的选择性吸附[70]。2018年,朱广山课题组制备的核-壳材料H5PMo10V2O40@ZIF-8@ZnO可用于制备传感装置,此装置在室温(25℃)下对甲醛(HCHO)具有改善的气体传感能力,灵敏度达到4×10-7(V/V)。另外,该材料对小尺寸的可还原气体分子表现出选择性响应,特别是甲醛相对于乙醇的选择性值为15.0[67]。
另外,Dolbecq课题组研究了POMs@UiO-67材料(POMs=[PW12O40]3-,[PW11O39]3-和[P2W18O62]6-)的电化学性质[56]。兰亚乾课题组制备了H5PMo10V2O40@HKUST-1与石墨烯复合得到的超级电容器[51]。王恩波课题组将[CoW12O40]@MIL-101(Cr)煅烧后制得的CoW12·Cr2O3@TiO2材料用于改善量子点太阳能电池的性能[58]。
4 结论与展望
近年来,化学家们成功利用原位合成、浸渍合成和机械研磨合成等方法,将POMs封装到多孔的MOFs(COFs)材料中,构筑了系列POMs@MOFs(COFs)材料。在该类材料中,POMs作为活性客体分子均匀分布在MOFs(COFs)材料中,既提高了MOFs(COFs)材料的稳定性又赋予其优异的性能,在光/电催化水分解制备清洁能源、制备生物柴油、燃油脱硫、化学战剂分解、传感器件以及电极修饰材料等领域具有潜在的应用前景。
目前制备的POMs@MOFs(COFs)材料中,作为客体的POMs以饱和的Keggin型结构为主,这主要得益于其高稳定性,而其它类型包括缺位的POMs在框架材料中装载的研究明显不足。作为装载主体的框架材料也主要以MOFs为主,COFs为装载主体的多酸基框架材料的研究则刚刚起步。而POMs@COFs型材料在催化以及传感器件等领域展示出巨大的应用前景,有待科学家们广泛深入研究。多酸基主客体框架材料在众多领域的广泛应用前景已引起化学家们的高度重视,并成为国际热点研究领域之一。我们期待多酸基主客体框架材料的合成及应用研究不断取得新的进展。
