固相萃取-超高效液相色谱-串联质谱法测定蜂产品中10种头孢类药物
2019-11-07吴银良张宜文许秀琴
章 豪, 吴银良, 张宜文, 许秀琴, 徐 峰
(1.宁波市农业科学研究院, 农业农村部农产品质量安全风险评估实验室(宁波), 浙江 宁波 315040;2.北京市计量检测科学研究院, 北京 100012)
头孢类药物属于广谱抗生素,临诊应用的头孢类药物均为半合成物质,杀菌力强,较青霉素类稳定,目前已发展到第五代[1]。然而过量使用头孢类药物会对人体健康带来潜在的危害,对儿童、老人,特别是胎儿的影响更大,严重者会导致胎儿病变、畸形、流产甚至死亡[2,3]。由于细菌产生耐药性和过敏反应等原因,目前我国已制定了头孢类药物的最高残留限量:头孢氨苄在牛肌肉、肝、肾、脂肪和奶中的最高限量分别为200、200、1 000、200和100 μg/kg;头孢喹肟在牛猪肌肉、肝、肾、脂肪和牛奶中的最高限量分别为50、100、200、50和20 μg/kg;头孢噻呋在牛猪肌肉、肝、肾、脂肪、牛奶中的限量分别为100、200、600和200 μg/kg,其中头孢噻呋的残留量以其代谢产物呋喃甲酰基头孢噻呋检测含量计算[4]。但目前头孢类药物在蜂产品中暂无限量规定。
目前对包括头孢类药物在内的β-内酰胺类药物残留分析的方法主要有液相色谱-串联质谱法(LC-MS/MS)[5-18]、高效液相色谱法(HPLC)[19-24]、毛细管电泳法[25,26]和微生物法[27]。20世纪90年代以后,绝大多数生物有机体中的β-内酰胺类药物残留分析采用液相色谱进行测定。然而使用液相色谱法测定时存在的主要问题是未经衍生化测定时,基体干扰较大;而经衍生化测定后灵敏度虽有所提高,但也只能作为一种筛选方法进行使用,对于分析多种头孢类药物仍存在极大难度。而超高效液相色谱-串联质谱法(UPLC-MS/MS)是一种集多组分定性、定量和高效分离于一体的系统,用于分析蜂产品中头孢类药物不仅能够显著提高方法的灵敏度和定性的准确度,而且能够降低前处理的成本和工作量[28]。而不论是采用HPLC还是UPLC-MS/MS方法测定,通常都采用固相萃取小柱进行样品的净化和富集[6,8,13]。根据头孢类药物的相关报道[29,30],考虑到药物的理化特性,前处理中净化一般选用Oasis HLB固相萃取小柱,且净化过程往往需要活化、淋洗和洗脱等费时费力的步骤。近来,Oasis PRIME HLB固相萃取小柱简单快速的通过式净化方案被用于快速有效地去除食品基质中的各种主要干扰物,包括脂肪、磷脂以及色素等[28]。Oasis PRIME HLB固相萃取小柱省去传统的活化、平衡步骤,样品经提取后直接过柱,这种样品净化方法简单、快速、有效,适合大批量日常分析检测,但目前并未发现有相关文献报道采用Oasis PRIME HLB固相萃取小柱用于蜂产品抗生素的样品前处理。
本研究采用Oasis PRIME HLB固相萃取小柱,通过对样品前处理方法和仪器条件进行优化,建立了基于UPLC-MS/MS测定3类主要蜂产品(蜂蜜、蜂王浆和蜂王浆冻干粉)中头孢类药物残留的分析方法。该方法检测周期短,准确度和精密度高,能满足多种蜂产品样品中头孢类药物的检测需要。
1 实验部分
1.1 仪器、试剂与材料
UPLC XevoTMTQ-S micro超高效液相色谱-串联质谱仪,配有电喷雾离子源(ESI)及MassLynx V4.1数据处理系统(美国Waters公司);低温高速离心机(德国Sigma公司); Vortex Genie 3漩涡振荡器(德国IKA公司)。
头孢类药物标准品(头孢匹林、头孢哌酮、头孢唑林、头孢乙腈、头孢拉定、头孢氨苄、头孢洛宁、头孢喹肟、去乙酰基头孢匹林、头孢噻肟,纯度均大于95%,德国Dr.Ehrenstorfer公司);甲酸(色谱纯,美国Tedia公司);乙腈、甲醇(色谱纯,德国Merck公司); Oasis PRIME HLB固相萃取小柱(500 mg/6 mL,美国Waters公司);实验用水为Milli-Q超纯水。
1.2 实验条件
1.2.1标准溶液配制
分别准确称取适量(精确至0.1 mg)10种头孢类药物标准品,用30%(v/v)乙腈水溶液溶解,配制质量浓度为1 g/L的标准储备液,于-20 ℃ 保存;分别准确吸取1 mL标准储备液,置于100 mL容量瓶中,用30%(v/v)乙腈水溶液稀释,配制质量浓度为10 mg/L的混合标准储备液,于-20 ℃保存。
基质匹配标准溶液:采用1.2.2节样品处理方法制备空白蜂产品样品溶液,稀释混合标准储备液,制备基质匹配标准溶液。
1.2.2样品处理
称取蜂蜜样品约5.00 g、蜂王浆样品约2.50 g、蜂王浆冻干粉样品约1.00 g(精确至0.01 g),分别置于50 mL离心管中,分别加入乙腈-水(80∶20, v/v)溶液20 mL,涡旋振荡1 min。蜂蜜样品直接移取溶液,蜂王浆、蜂王浆冻干粉样品以9 500 r/min高速离心5 min,移取上清液至Oasis PRIME HLB固相萃取小柱,收集全部流出液,流出液在40 ℃下氮气吹干;用1 mL 0.1%(v/v)甲酸水溶液-甲醇(95∶5, v/v)溶液溶解,涡旋振荡,过0.22 μm滤膜,用于UPLC-MS/MS分析。
1.2.3色谱条件
色谱柱:Acquity UPLC BEH C18(100 mm×2.1 mm, 1.7 μm);流动相:A相为0.1%(v/v)甲酸水溶液,B相为甲醇;梯度洗脱程序:0~1.0 min, 5%B; 1.0~4.5 min, 5%B~50%B; 4.5~6.0 min, 50%B; 6.0~6.1 min, 50%B~5%B; 6.1~7.5 min, 5%B;流速0.30 mL/min;进样量10 μL。
1.2.4质谱条件
多反应离子监测模式(MRM); ESI源正离子模式电离;脱溶剂气流速:1 000 L/h;锥孔气流速:50 L/h;离子源温度:150 ℃;脱溶剂气温度:500 ℃;毛细管电压:2.0 kV; RF透镜电压:0.5 V;萃取锥孔电压:25 V;头孢类药物的相关参数见表1。
2 结果与讨论
2.1 仪器条件优化
基于LC-MS/MS测定头孢类药物残留大多采用ESI源正离子模式电离的分析方法[5-18]。本实验采用甲醇-水(5∶95, v/v)溶液分别配制10种头孢类药物标准溶液(2.0 mg/L),利用超高效液相色谱-串联质谱仪自带的Intellistart软件分别获得10种头孢类药物的母离子、子离子、锥孔电压和碰撞能量(见表1),发现它们在正离子模式下均获得了较好的响应。

表1 10种头孢类药物的母离子、子离子、锥孔电压和碰撞能量
* Quantitative ion.
本实验考察了乙腈和甲醇作为色谱流动相的有机相时头孢类药物的分离效果,发现当采用乙腈作为有机相时,头孢哌酮、头孢乙腈等头孢类药物的响应较差,而采用甲醇作为有机相时,10种药物的响应均较好,因此确定甲醇为有机相。
2.2 前处理条件优化
生物样品中头孢类药物的常用溶剂有甲醇[5]、甲醇-水[9]和乙腈-水[5,6,13]等。因蜂产品特别是蜂王浆和蜂王浆冻干粉样品中通常含有蛋白质和磷脂类物质,用甲醇或甲醇-水溶解时样液较混浊,需进一步净化去除蛋白质。而用乙腈溶解时,因蛋白质变性沉淀,溶解后杂质少,且溶剂澄清,根据相关文献报道,目前用于动物性食品溶解的乙腈溶液中乙腈含量均高于60%[5,12,19],因此本研究使用乙腈、乙腈-水(80∶20, v/v)和乙腈-水(60∶40, v/v)3种溶剂对蜂产品在200 μg/kg加标水平下进行溶解上样,结果发现采用乙腈-水(60∶40, v/v)上样时,不能使所有头孢类药物的回收率达到75%以上;而采用乙腈-水(80∶20, v/v)上样时,所有头孢类药物的回收率均大于75%;采用乙腈上样时,所有头孢类药物的回收率大于75%,但考虑到乙腈的强洗脱能力,样品以及小柱中所含的杂质也会一起被洗脱下来,失去了过柱净化的目的,因此确定以乙腈-水(80∶20, v/v)进行溶解上样。
2.3 固相萃取小柱的选择
本实验对Oasis PRIME HLB和Oasis HLB两款固相萃取小柱进行了比较。我们在蜂产品中选取最为主要的蜂蜜、蜂王浆和蜂王浆冻干粉3类基质进行试验,从每一类基质中选取代表样品进行添加回收试验,其中,蜂蜜中头孢哌酮、头孢乙腈和头孢唑林3种头孢类药物的添加水平为4、20、200 μg/kg,其余7种头孢类药物的添加水平为1、20、200 μg/kg;蜂王浆中头孢哌酮、头孢乙腈和头孢唑林3种药物的添加水平为8、40、200 μg/kg,其余7种头孢类药物的添加水平为2、20、200 μg/kg;蜂王浆冻干粉中头孢哌酮、头孢乙腈和头孢唑林3种药物的添加水平为20、50、200 μg/kg,其余7种头孢类药物的添加水平为5、50、200 μg/kg。结果表明,样品采用Oasis PRIME HLB固相萃取小柱净化后的回收率(75.0%~89.8%)均优于采用Oasis HLB固相萃取小柱净化的结果(60.0%~71.0%)。结果还显示,在无需活化、淋洗和洗脱等复杂的步骤下,样品采用Oasis PRIME HLB固相萃取小柱净化后,无论是在回收率还是基质干扰方面,均优于采用Oasis HLB固相萃取小柱净化的结果。这可能是因为对于蜂产品特别是蜂王浆和蜂王浆冻干粉中的磷脂和蛋白质等杂质,相比Oasis HLB固相萃取小柱,Oasis PRIME HLB固相萃取小柱可以更好地吸附这些杂质,不仅无需活化、淋洗和洗脱等这些费时费力的前处理步骤,在显著缩短净化时间的同时还能延长色谱柱的使用寿命[31,32]。因此本实验采用Oasis PRIME HLB固相萃取小柱作为净化小柱。
2.4 基质效应、标准曲线、检出限和定量限
基质效应主要是由于样品在离子化时基质成分与目标化合物相互竞争电离所致,包括基质增强效应和基质抑制效应。在UPLC-MS/MS定性定量分析中,基质效应会对仪器的灵敏度和重复性产生影响,进而影响检测结果的准确性。本实验采用(基质匹配标准曲线的斜率/溶剂标准曲线的斜率)×100%的计算方法对10种头孢类药物在蜂产品基质中的基质效应进行考察。当结果大于100%时表示存在基质增强效应,小于100%时表示存在基质抑制效应,结果越接近100%基质效应越小。由表2可以看出,蜂产品有明显的基质增强作用,10种头孢类药物均存在不同程度的基质增强效应。为了能更准确地测定目标化合物,本实验采用基质匹配标准曲线来消除基质效应。采用蜂产品空白基质液配制1~1 000 μg/L横跨3个数量级的系列基质标准工作溶液(其中头孢乙腈、头孢哌酮和头孢唑林为5~1 000 μg/L)进行UPLC-MS/MS测定分析,以峰面积对相应质量浓度作标准曲线,得到线性回归方程和相关系数(r2),如表2所示。10种头孢类药物在相应浓度范围内的r2均大于0.999,线性关系良好。按3倍信噪比(S/N=3)计算,10种头孢类药物的检出限(LOD)为0.15~1.5 μg/kg;按S/N=10计算,10种头孢类药物的定量限(LOQ)为0.50~5.0 μg/kg。农业部235号公告规定头孢喹肟、头孢氨苄在动物性食品中的最大残留限量为20~1 000 μg/kg,而本方法检出限低至0.15~1.5 μg/kg,因此能满足蜂产品中头孢类药物含量分析的需要。
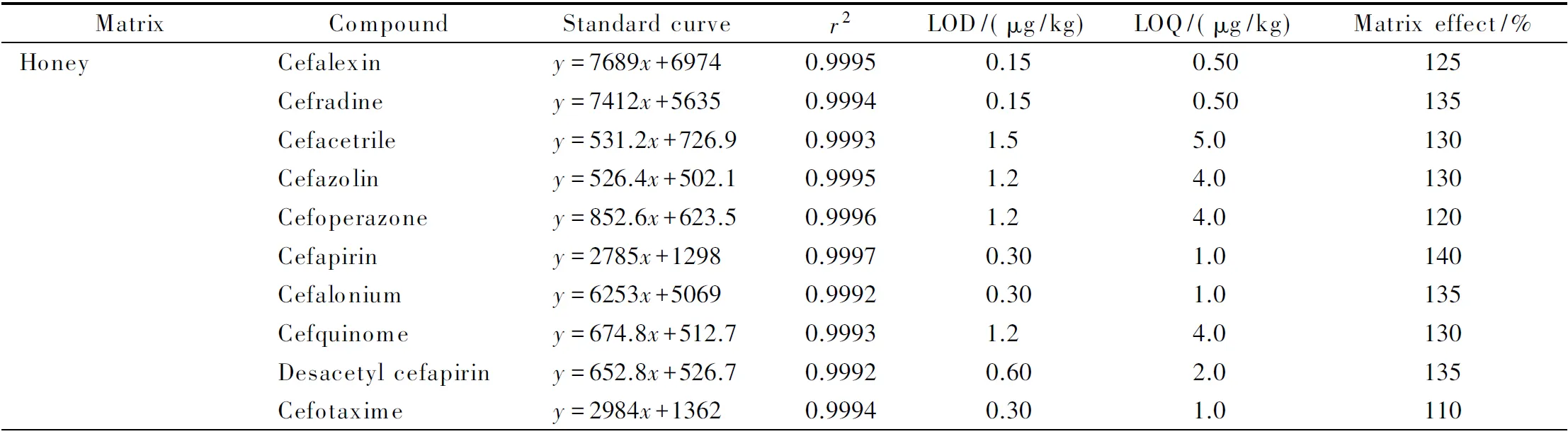
表2 10种头孢类药物在不同基质中的标准曲线、相关系数、检出限、定量限和基质效应

表2 (续)
y: peak area;x: mass concentration, μg/L.
2.5 回收率与精密度
选取蜂蜜、蜂王浆和蜂王浆冻干粉3类基质进行添加回收试验,计算相对标准偏差(RSD),结果见表3。头孢类药物在蜂蜜空白样品中的加标MRM色谱图见图1。蜂蜜中头孢哌酮、头孢乙腈和头孢唑林的添加量为4 μg/kg,其余7种头孢类药物的添加量为1 μg/kg,每批次同一水平5个平行样品,重复测定3次。
由表3可知,10种头孢类药物的加标回收率为75.0%~89.8%,RSD为1.4%~4.6%。上述结果表明,该方法对测定蜂蜜、蜂王浆和蜂王浆冻干粉中10种头孢类药物的残留均具有较好的准确度和精密度。
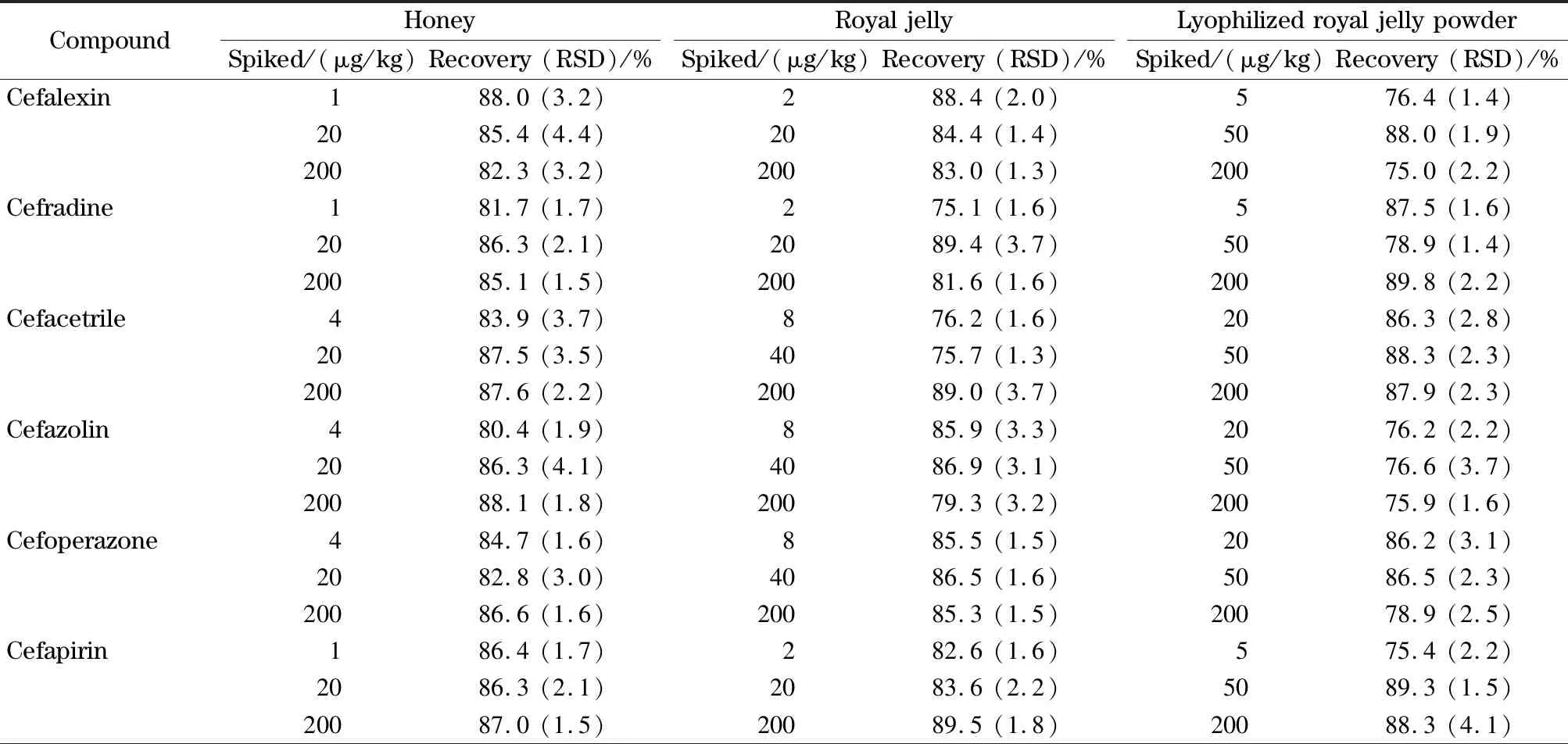
表3 10种头孢类药物在蜂蜜、蜂王浆、蜂王浆冻干粉中的回收率和相对标准偏差(n=5)
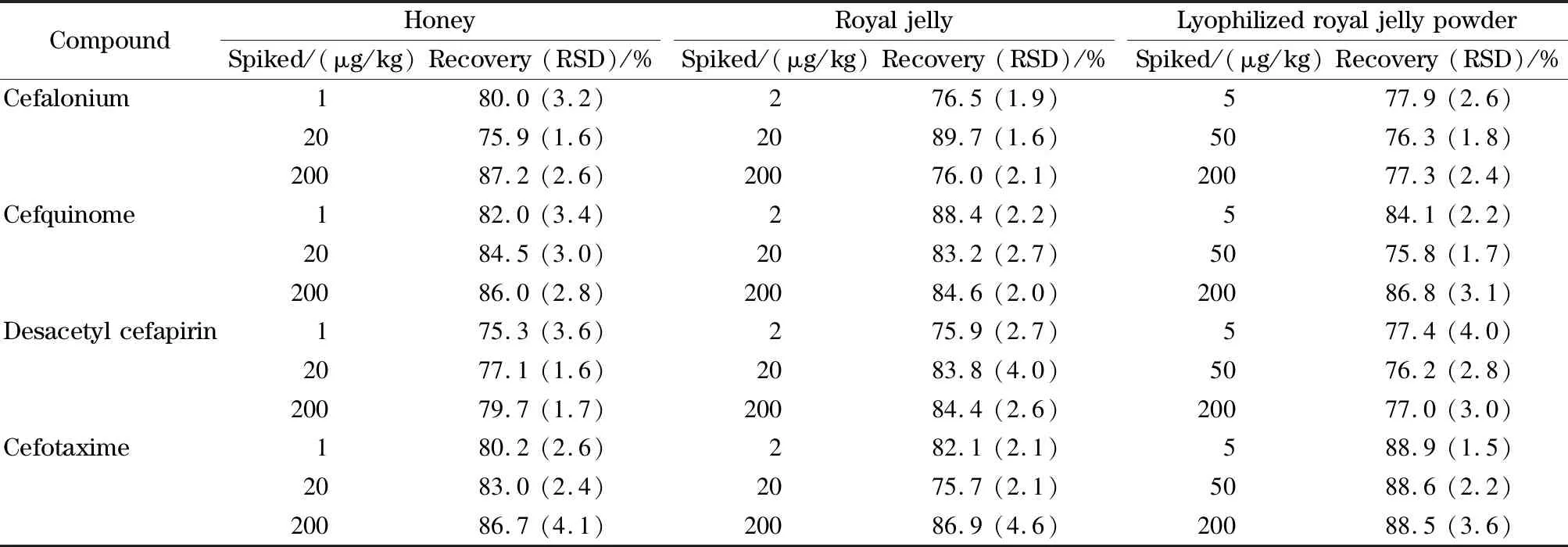
表3 (续)
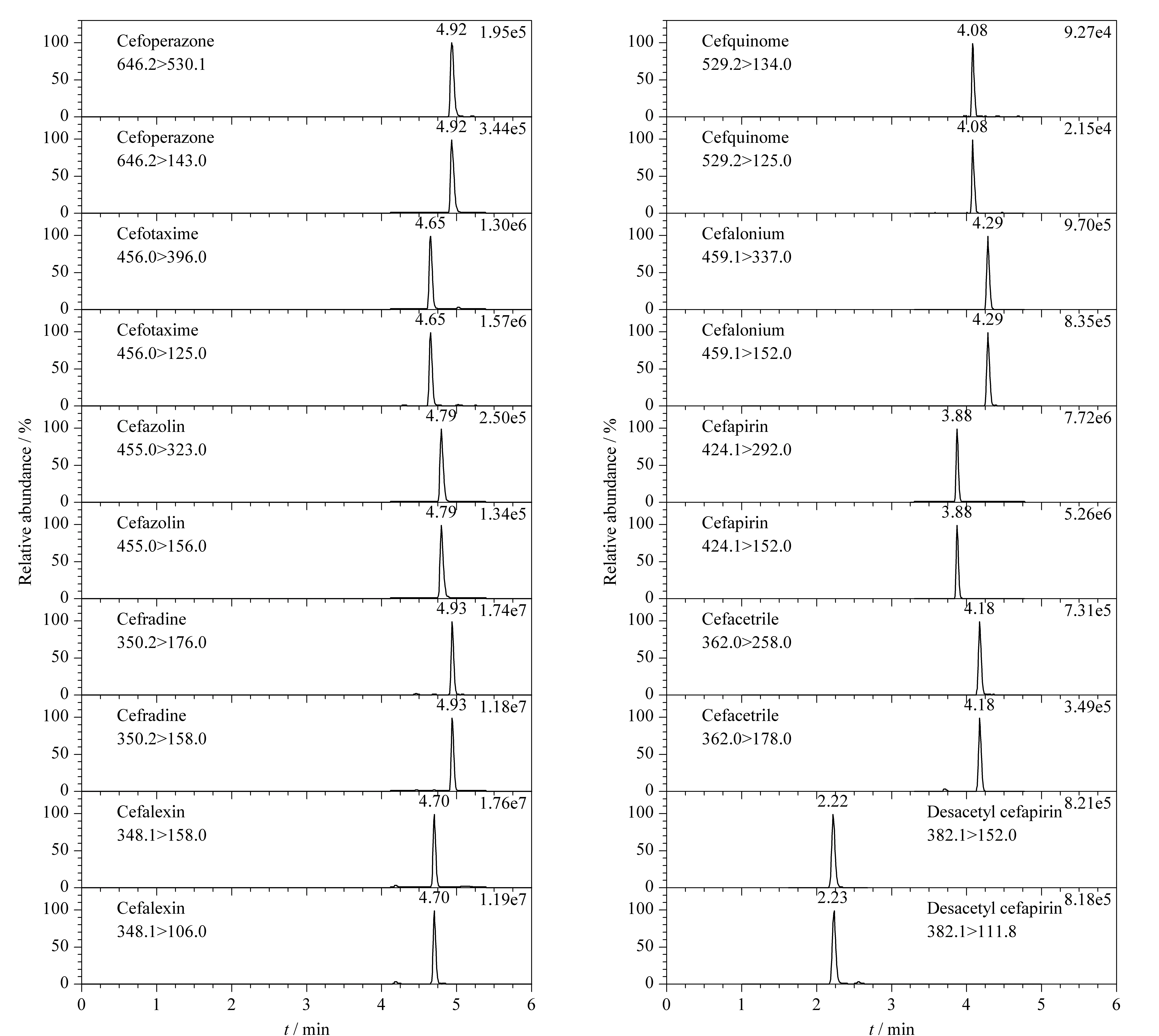
图1 蜂蜜加标样品的MRM色谱图
2.6 方法应用
采用优化后的仪器条件和前处理条件分别对随机采集的50个蜂产品(含蜂蜜、蜂王浆和蜂王浆冻干粉)样品进行分析,均未检出上述10种头孢类药物。我们参照GB/T 22942-2008《蜂蜜中头孢唑啉、头孢匹林、头孢氨苄、头孢洛宁、头孢喹肟残留量的测定 液相色谱-串联质谱法》对实际样品进行了复测,结果一致。
3 结论
本工作建立了同时分析蜂产品中10种头孢类药物的超高效液相色谱-串联质谱方法,样品经乙腈-水(80∶20, v/v)溶液提取后离心,上清液经Oasis PRIME HLB固相萃取柱净化,氮吹后复溶进样分析。方法具有精密度和准确度高、检测周期短的优点,能满足蜂产品中10种头孢类药物含量分析的需要。
