B细胞双向调控在系统性红斑狼疮致病和治疗中的研究进展*
2019-11-04张乃丹段思雨喻敬芯武永康
张乃丹,段思雨,喻敬芯,武永康△
1.四川大学华西医院 实验医学科(成都 610041); 2.德阳市人民医院 检验科(德阳 618000)
系统性红斑狼疮(systemic lupus erythematosus,SLE)发病过程是由多因素调控的渐进过程,分为亚临床期、急性炎症期、慢性自身免疫病期和转归期4个阶段[1](图1)。B细胞作为SLE发病机制中的重要环节,其表面表达很多免疫激活和免疫抑制作用的分子。激活分子包括B细胞受体(B cell receptor, BCR)[2]、Toll样受体(Toll-like receptor,TLRs)[3]和B细胞表面抗原CD40[4]等。抑制分子包括Fc受体FcγRIIB (Fcγ receptor IIB, FcγRIIB)[5]和CD72[6]、程序性死亡蛋白1(programmed death 1 protein,PD-1)[7]等,这些分子所提供的信号作用于B细胞, 影响B细胞活化的性质和程度,控制由此产生的B细胞亚群分化及抗体产生[8](图2)。B细胞表面分子双向调控失衡可导致B细胞免疫功能出现异常,进而导致SLE发生。鉴于B细胞靶向治疗越来越多地应用于临床,充分理解B细胞表面分子在SLE炎症调节的不同作用,并设计出安全有效的免疫治疗方案可能是今后研究的重点。本研究综述了B细胞正向负向两类调控分子及相关靶向治疗在SLE中的研究进展,充分认识不同分子对B细胞功能的调控在SLE发病机制中的作用及靶向治疗中的应用,为SLE治疗药物设计提供新的策略。
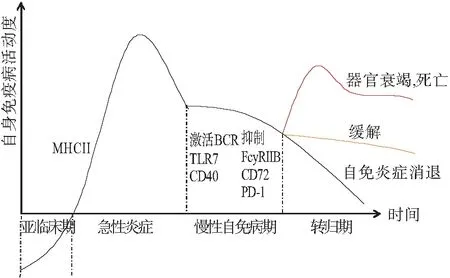
图1 SLE疾病分期及正向负向B细胞调节分子
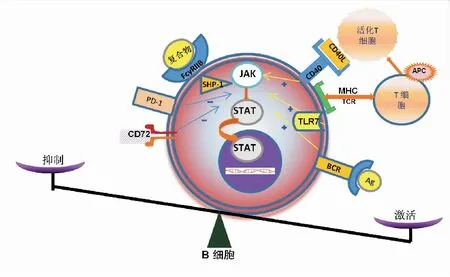
图2 B细胞双向调节及B-T细胞相互作用与SLE免疫平衡的关系
1 B细胞激活因素
1.1 BCR
SLE与BCR信号传导的异常活化密切相关,激活BCR信号通路与Ca2+信号及其调节受体和适配器密切相关[2]。酪氨酸激酶介导酪氨酸免疫受体激活基序(immunoreceptor tyrosine based activation motif, ITAM)磷酸化,激活促进BCR通过连接B细胞链接蛋白(B-cell linker protein, BLNK)进行信号传递。BCR介导磷酸化磷脂酶C-γ2(phospholipase Cγ2, PLCγ2)切割膜相关磷酰氨基肽(phosphatidylinositol-4,5-bisphosphate,PIP2)为1,4,5三磷酸肌醇(Inositol 1,4,5-trisphosphate, InsP3)和二酰基甘油(diacylglycerin,DAG)。InsP3引起细胞内外Ca2+信号活化进而激活核因子(nuclear factor-κB,NF-κB)。DAG调节激活有丝分裂激活蛋白激酶(phosphorylated mitogen-activated protein kinase, MAPK)、c-Jun NH2末端激酶(c-jun-N-terminal kinase, JNK/SAPK)和p38 MAPK。在BCR调控的Ca2+信号失衡在SLE发病机制中的研究中发现[9],对于敲除特异性B细胞中SH2酪氨酸磷酸酶1[Src homology 2 (SH2) domain containing protein tyrosine phosphatase-1, SHP-1]的CD19-Cre 小鼠,SHP-1缺失增强了BCR激活Ca2+信号,加速了SLE的活动,其特征为产生抗dsDNA抗体和导致肾小球肾炎的发生。关于SLE患者B细胞直接磷酸化研究[10]发现,BCR激活后细胞内钙释放减少,SLE患者B细胞中丝氨酸和酪氨酸磷酸酶失衡有助于BCR激发促进SLE患者B细胞的分化产生浆细胞,继而促进自身抗体的产生。
1.2 TLR7
TLR7是一种Ⅰ型跨膜蛋白,是已知SLE疾病模型的致病因素之一。TLR7识别单链RNA和鸟苷酸类似物,通过B细胞内C端区域激活下游信号通路。TLR7发生构象变化识别配体,适配器蛋白被招募到Toll/白细胞介素-1受体(Toll/interleukin-1 receptor,TIR)域。TIR域启动信号级联激活NF-κB或干扰素调节因子(interferon regulatory factor,IRF),促进编码NF-κB/IRF的mRNA转录[11]。TLR7相关SLE样模型表现为染色质含量和调控分子的异常。Fairhurst等[12]研究显示,BXSBYaa小鼠自发诱导Y染色体上增加了TLR7基因的复制。BXSBYaa小鼠同时表达了X和Y染色体上2份TLR7的拷贝,对内源性配体反应增强,表现为促炎作用增强。同时TLR7的控制分子对于调控炎症也很重要。Unc93同源B1 (Unc93 homolog B1, UNC93B1)是TLRs核酸敏感性的重要分子之一,UNC93B1与TLRs结合将其运送到内质网,通过细胞外域的蛋白质分解促进TLRs成熟。一项来自埃及开罗大学的研究[13]显示,TLR7基因型(rs385389)和狼疮肾炎显著关联。关于波兰SLE患者的研究[14]发现,TLR7基因(rs1634318和rs1616583)与SLE相关,但与盘状红斑狼疮不相关。有研究[15]表明,女性复等位表达TLR7的B细胞表现出更高促进SLE炎性反应的功能。
1.3 CD40
2 B细胞抑制因素
2.1 Fc受体FcγRIIB
Fc受体(Fc receptors, FcRs)在结构上有同源的Ig结构域,但功能上只存在激活型和抑制型。使用FcRs缺陷小鼠研究[18]表明,敲除小鼠FcγRIIB导致SLE样改变,骨抗弯强度和硬度降低导致骨质疏松症和骨折风险增加。FcγRIIB是含有酪氨酸免疫受体抑制基序(immunoreceptor Tyrosine based inhibitory motifs,ITIM)的FcRs,当通过免疫复合物与激活受体聚集时,会触发B细胞负性调节信号。ITIM被磷酸化,磷酸化的FcγRIIB具有高度亲和性的Src同源2(Src homology 2, SH2)结构域,大量的SHP-1被招募并被带入激活FcRs产生的信号体,进一步阻断下游信号的激活[19]。来自麻省总医院的研究[5]显示,跨膜(transmembrane, TM)域内单核苷酸多态性(rs1050501)具有阻断FcγRIIB-I232T的功能,并且与SLE易感性相关联,由此也表明Fc受体FcγRIIB抑制信号分子对SLE的潜在保护作用。
2.2 CD72
CD72是一种45 kDa 的Ⅱ型跨膜糖蛋白,作为BCR的共受体在B细胞上表达,在降低B细胞过度活化方面发挥着重要作用。CD72在B细胞上正常表达时,CD72在成熟B细胞中下调BCR下游的主要信号转导通路,包括NF-AT、NF-κB、JNK和胞外信号调节激酶(extracellular signal-regulated kinase,ERK)。在成熟B细胞中的作用表现为促进细胞分裂周期阻滞,改变B细胞增殖活化,促进抗原刺激后程序性细胞死亡。Vadasz等[20]研究发现,CD72在SLE患者外周血显著增加,并与肾脏受累相关。可溶性CD72可能成为在SLE肾脏受累潜在生物标志物。另有研究[21]发现,SLE患者活化的B细胞表面CD72表达较正常人显著降低,与SLE疾病活动性和补体水平负相关。CD72作为SLE患者B细胞的抑制性分子,其机制在于它能够识别Sm/RNP (Sm/Ribonucleoprotein, RNP),并通过激活SHP-1抑制Sm/RNP反应性B细胞活化。CD72参与抑制TLR7介导的RNA在核蛋白复合物中对核酸酶产生反应,促使游离RNA在遇到核内RNA传感器前被核酸酶降解进而阻断TLR7信号通路而发挥抑制作用。
2.3 PD-1
程序性死亡受体-1(programmed death 1,PD-1)属于CD28/CTLA-4家族,PD-1通过与配体PD-L1(CD 274)和PD-L2(CD 273)结合产生抑制信号,降低免疫介导的组织损伤。在SLE患者相关研究[22]发现,CD19+PD-L1+的B细胞在SLE病人的外周血中表达异常,且疾病相关实验室指标和临床特征相关,预示了CD19+PD-L1+的B细胞在SLE发病机制中起到关键作用。在NZB / NZW F1小鼠动物模型中研究[23]表明,PD-1 靶向药物作用于CD28,进一步减少B细胞活化和抗dsDNA抗体的产生,减缓了狼疮肾炎的进展。
3 B细胞和T细胞相互作用与相关靶向治疗
B细胞双向调节分子及B-T细胞相互作用可精确调控SLE的平衡状态。异常细胞因子、趋化因子及生长因子的产生、包括T细胞免疫应答中性粒细胞胞外诱捕网(neutrophil extracellular traps, NET)相关分子的清除缺陷、血浆中异常外切体释放,干扰素α过度表达等因素促使T细胞朝向Th2和Th17细胞途径分化,并导致多克隆B细胞活化,增加程序性细胞死亡和NET形成。T细胞活化产生IL-4、IL-21,加速激活B细胞以及自身抗体产生。先前研究[2-4]评估了BCR交联、sCD40L、TLR和细胞因子刺激后控制B细胞分化的基因调控网络的变化,发现在B细胞激活的3个信号中,通过激活酪氨酸激酶,如Syk/Btk/JAK,结局直接导致滤泡辅助性T细胞(follicular helper T cells, Tfh)、炎症因子和自身抗体的产生。与健康人相比,SLE患者B细胞Syk/Btk磷酸化水平明显升高。在SLE患者中,酪氨酸激酶如Syk/Btk/JAK是抑制B-T细胞相互作用的潜在靶点,目前的靶向药物阿巴西普(Abatacept)是CTLA-4分子的细胞外功能区与人IgG1的Fc段结合而成的可溶性融合蛋白,该药物治疗患者可显著降低外周血B细胞中p-Syk的水平,同时降低Tfh细胞的比例[24]。
淋巴信号活化家族 (signaling lymphocyte activation molecule family, SLAMF)中的受体CS1(CD319, SLAMF7)是一种自身配体,通过增加自分泌细胞因子的产生,诱导B淋巴细胞增殖。在一组基于SLE家族关联研究[25]的结果显示,在SLE患者的家族中,CS1基因存在突变,增加了SLE疾病发生的风险。CS1的重要特征之一是这些受体本身可作为自配体与其他免疫细胞上的CS1相互作用。SLE患者循环的B细胞有不同的亚群,如记忆B细胞、幼稚B细胞和浆细胞[26]。Kim等[27]研究结果显示,SLE患者外周血单核细胞中CD19阳性B细胞的不同亚群可进一步根据CS1表达水平进行分类。CS1较高水平表达有利于B细胞增殖,而CS1在B细胞上表达是由CD40介导的B细胞活化诱导产生的。同时表明SLE疾病活动与高频率浆细胞和自反应记忆B细胞有关。另一组研究[28]中比较了SLE患者和健康对照者外周血T细胞,B细胞及其各自分化亚群的SLAMF1-7的表达,结果显示SLE患者的T/B细胞及其分化亚群的SLAMF1水平均升高。SLAMF4和SLAMF7在SLE患者CD8 T细胞的表达降低。SLAMF3在SLE CD4和CD8 T细胞表达略有增加。在SLE中各种SLAMF受体表达失调,有助于相关免疫疗法的研究。目前受到广泛关注的嵌合抗原受体-T细胞(chimeric antigen receptor engineered T cells, CAR-T)正是利用了CS1的表达在SLE中发生了改变,但围绕CAR-T细胞的研究和以CS1为靶向治疗SLE还需要进一步临床药物试验明确其有效性和安全性。更好地理解CS1的作用将使我们能够在自身免疫性疾病的免疫治疗中利用这些靶向受体。临床医师虽然渴望更有针对性的治疗药物出现,但同样关注可能存在的药物不良反应,这需要更长时间的观察和研究,特别是实验动物模型研究成果如何指导SLE患者临床治疗是急需解决的主要问题。
4 结语与展望
随着人们对SLE疾病分期、慢性SLE疾病期B细胞中双向调节以及B-T细胞间相互作用的深入研究,更多关于B细胞为核心的促炎/抑炎的靶向分子将会成为研究热点。尽管目前对于SLE的B细胞信号通路的靶向药物仍处于临床药物试验阶段,但我们坚信针对B细胞双向精准调控的免疫制剂将会为SLE的治疗提供更多的策略。
