Transcriptional addiction in mixed lineage leukemia: new avenues for target therapies
2019-11-02RuijingXioHonghongWngKiweiLing
Ruijing Xio, Honghong Wng, Kiwei Ling,c,*
aDepartment of Pathophysiology, School of Basic Medical Sciences, Wuhan University, Wuhan, P.R.China; bDepartment of Immunology, School of Basic Medical Sciences, Wuhan University, Wuhan, P.R.China; cResearch Center for Medicine and Structural Biology, School of Basic Medical Sciences, Wuhan University, Wuhan, P.R.China
Abstract
Keywords:DOT1L complex, Leukemogenesis, Mixed lineage leukemia (MLL), MLL chimeras, Superelongation complex (SEC),Transcriptional addiction
1.HISTORICAL OVERVIEW OF MLL LEUKEMIA
Acute leukemia is a class of malignant blood cancers arising from the hematopoietic stem and progenitor cells (HSPCs); it is generally characterized by recurring chromosomal translocations,mutations,and epigenetic alternations.The diagnosis and classification of acute leukemia mainly relies on multidisciplinary approaches, such as bone marrow biopsy, karyotype analysis,molecular genetic testing, and immunophenotyping, for categorization into myeloid,B-lymphoid,or T-lymphoid lineage.In the 1980s,when monoclonal antibodies were applied to characterize the leukemic blasts,three acute myeloid leukemia(AML)patients were noted to co-express myeloperoxidase and terminal deoxynucleotidyl transferase, which had been discovered to be a biochemical marker for acute lymphoblastic leukemia (ALL).Such findings indicate the presence of leukemic blasts with both lymphoid and myeloid markers.1In 1985, St.Jude Children's Research Hospital reported the clinicopathological features of a cohort of 123 children leukemia with lymphoid and myeloid characteristics using lymphoid-associated markers,such as anti-CD10,CD2,and CD5,and myeloid-associated markers,such as CD15, CD13, and CD11b,2although these markers are no longer considered to be lineage specific.Based on these markers,a subset of these children leukemia(25/123)were characterized to have individual blasts expressing differentiation markers of more than one lineage;thus the term“mixed lineage leukemia”(MLL)was initially defined.
Since the 1980s,cytogenetic studies3-5on acute leukemia have revealed that 11q23 translocations are the most common genetic alteration in infants with leukemia.6These translocations are linked with the mixed-lineage characteristics by affecting progenitor cells capable of both myeloid and lymphoid differentiation at early stages.7,8In 1991, Janet Rowley's group first identified a gene spanning a breakpoint in the 11q23 translocations and named it MLL, meaning myeloid/lymphoid,or mixed-lineage,leukemia9;this gene was cloned and sequenced by another three groups,10-12who also showed that the MLL(KMT2A)gene is the homolog of Drosophila trithorax.In 1995,the Mll-/-mice were reported to be embryonic lethal, whereas Mll+/-mice were found to be haploinsufficient with segmentation defects and hematological abnormalities.These findings suggest that MLL plays a critical and nonredundant role in development and hematopoiesis.13MLL has also been shown to be essential for adult hematopoietic stem cell self-renewal.14,15
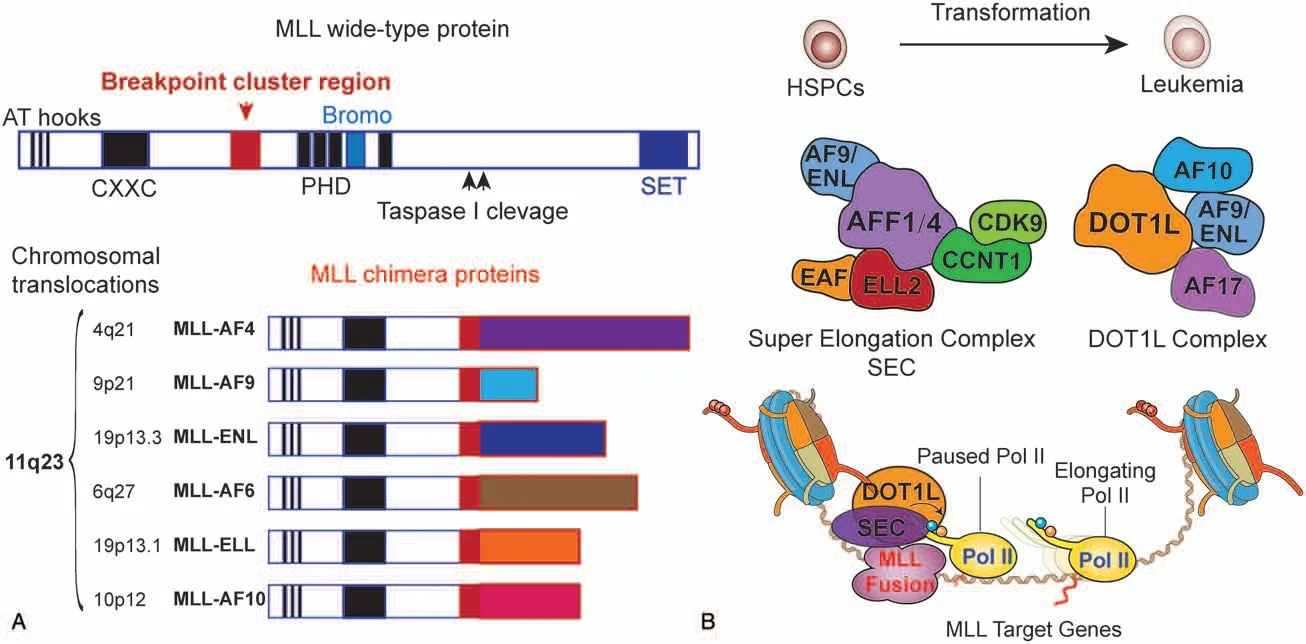
Figure 1.Transcriptional misregulation of MLL chimeras,SEC,and DOT1L complexes in MLL leukemia.(A)MLL is a multidomain protein with approximately 4000 amino acids and contains three AT-hook domains,a CXXC domain that binds nonmethylated CpG islands,chromatin-binding PHD fingers and bromodomain,and a SET domain at the C-terminus of MLL.MLL breakpoints vary in different patients and locate mainly between exon 7 and exon 10,which was designated a region called“breakpoint cluster region”.The resulting MLL fusion proteins retain the chromatin-binding CXXC module.(B)Identification of SEC and DOT1L complexes containing the most common MLL fusion partners.Fusion of MLL and SEC/DOT1L subunits could aberrantly recruit SEC and DOT1L complexes to activate the MLL target genes and promote the transformation of HSPCs to leukemia.
Leukemia with 11q23 translocations has been demonstrated to feature MLL gene rearrangements,16and multiple partner genes were found to be fused with the MLL gene, resulting in diverse oncogenic MLL chimeras composed of an MLL N-terminal portion fused in-frame to a C-terminal fragment of the partners(Fig.1A).Based on an updated recombinome of MLL in 2017,17a total of 135 different MLL rearrangements have been identified and 94 fusion partner genes have been characterized at the molecular level.These fusion partners share few sequence or functional similarities, and the most common partner genes are AF9 (MLLT3),AF4 (AFF1),ENL (MLLT1),AF10 (MLLT10),ELL, AF6 (MLLT4), EPS15, and SEPT6.In 2002, Armstrong et al reported that MLL-rearranged leukemia is clearly different from conventional acute leukemia using gene expression profiles from microarrays; MLL leukemia has been reported to be a unique entity.18Interestingly, partial tandem duplication (PTD)of the MLL gene, which produces an extra amino-terminus inframe to the full-length MLL protein,has been found in AML and myelodysplastic syndrome (MDS) with unique characteristics.MLL-PTD has been thoroughly described in a recent study19and,therefore, will not be the discussed in this review.
2.CLINICAL FEATURES OF MLL LEUKEMIA
MLL translocations predominantly occur in pediatric AML and ALL,including 69%-79%of all infant ALL cases(aged <1 year) and approximately 10%of all other ALL cases,>90% of all infants with congenital ALL(aged <1 month),and 35%-50%of infants with AML.20MLL translocations also present less often in older children and adult patients with acute leukemia,especially AML,compared with their younger counterparts.21,22Patients treated with topoisomerase inhibitors can develop therapy-related or secondary leukemia; of these patients, 10%carry MLL translocations similar to de novo ALL and AML.22-24Considering the broad spectrum of the MLL recombinome and the fact that molecular genetic tests do not cover all MLL fusion genes, the incidences of MLL translations may be underestimated.
Compared with other subsets of acute leukemia, MLL leukemia is more likely to be associated with certain phenotypic features such as hyperleukocytosis, central nervous system infiltration, and therapy refractoriness.MLL leukemia is generally associated with dismal prognosis due to the development of resistance and high chance of relapse with established therapies, including chemotherapy and hematopoietic stem cell transplantation.25,26A recent study also showed that MLL leukemia could circumvent CD19-directed immunotherapies through lineage plasticity.27Adults who survive childhood leukemia tend to suffer long-term and multiorgan-related health effects.28As such,MLL leukemia remains a clinical challenge for which the development of more effective target therapies is urgently needed.29
3.EPIGENETIC AND TRANSCRIPTIONAL REGULATION BY MLL PROTEINS
The MLL gene is conserved with Drosophila trithorax and yeast Set1; it encodes a member of the complex of proteins associated with Set1 (COMPASS) family of enzymes,30which catalyze methylation of histone H3 lysine 4 (H3K4) via a conserved SET domain.31,32MLL is a large,multidomain protein with approximately 4000 amino acids; it can be extensively cleaved by Taspase I to generate an N-terminal 320-kDa fragment (N320) and a C-terminal 180-kDa fragment (C180)(Fig.1A).33The N320 fragment contains three AT-hook domains near the N terminus of MLL, a CXXC domain that binds nonmethylated CpG islands, PHD fingers, and a bromodomain that are chromatin-binding modules,as well as a conserved SET domain located at the C-terminus of the C180 fragment(Fig.1A).The N320 and C180 fragments interact together to form the MLL complexes with the association of other proteins such as WDR5, DPY30, ASH2L, RBBP5, MEN1, and HCF1.34
MLL rearrangements mostly occur at one allele of MLL gene and occur between the CXXC domain and the first PHD finger,resulting in inclusion of the chromatin-binding domain CXXC and loss of H3K4 methylation activity in the MLL chimeras.Conservation of MLL with Drosophila trithorax to maintain the expression of HOX genes10-12leads to two major hypotheses35:(1) MLL chimeras misregulate a subset of MLL target genes,36and, therefore, transform HSPCs to MLL leukemia through alternated transcriptional regulation and(2)HOX genes mediate leukemogenesis in MLL leukemia.Consistent with these two hypotheses,MLL leukemia has a highly distinct gene expression profile and is generally associated with high expression of the transcription factors HOXA9 and MEIS1,which are capable of transforming hematopoietic stem cells (HSCs) into leukemia cells.18,37However,MEIS1 is consistently expressed in leukemia with high expression of HOXA9,18,38and the knockout of Hoxa9 in Mll-AF9 knock-in mice does not affect the incidence of MLL leukemia.39Additionally, MLL chimeras could transform both HSCs and highly differentiated granulocyte-macrophage progenitors (GMPs), whereas HOXA9 and MEIS1 can only transform HSCs.40Moreover,HOXA9 and MEIS1 proteins are transcription factors functioning at the top of their regulatory cascade; the downstream targets of these proteins and their cooperating factors in MLL leukemogenesis and transcriptional regulation remain unknown.
4.COOPERATING EVENTS IN MLL LEUKEMIA
The retroviral transduction strategy and knock-in mouse models41,42have demonstrated that MLL chimeras can induce acute leukemogenesis.However, the long latency in knock-in mice and monoclonal nature of the induced leukemia indicate that multiple cooperating events or signaling pathways may be required to promote the pathogenic processes,43consistent with the “two-hit” or Knudson hypothesis44in cancer.Since 2000s,multiple signaling pathways, mediated by FLT3; RAS/MEK/ERK;Integrin β3/SYK;Wnt/β-catenin;GSK3;NFκB;CEBPα and PU.1; chromatin-modifying complex; and the target genes of MLL chimeras such as HOXA9, MEIS1, EVI1, MEF2C, CMYC, miR196b, and PBX proteins, have been reported to promote MLL leukemogenesis.35Here,we describe the relevant cooperating events,including BRD4,CBP/P300,WDR5,MEN1,CDK6,MSI2,JMJD1C,and CAF1 complex,and their potential use.
Bromodomain-containing protein 4 (BRD4) BRD4 interacts with acetylated histone 3 lysine 27 (H3K27) and promotes the transcription of MYC, and this interaction can be blocked by small-molecule BET domain inhibitors.45In 2011, an unbiased RNA interference screening46with chromatin modifiers identified BRD4 as a therapeutic target in MLL-AF9 leukemia through the downregulation of MYC expression by the BET domain inhibitor JQ-1.Two months later,Kouzarides's group47reported another BET domain inhibitor, I-BET151, as a potential compound for both MLL-AF9 and MLL-AF4 leukemia through the induction of early cell cycle arrest and apoptosis.BET domain inhibitors have been demonstrated to suppress the function of hematopoietic transcription factors in AML.48However, in 2015, two independent groups49,50reported the recurring development of resistance to BRD4 inhibitors through increased Wnt/β-catenin signaling and rewiring of transcriptional programs, which highlights the potential therapeutic limitations of BET inhibitors in MLL leukemia.
CREB-binding protein(CBP)and E1A-binding protein(P300),which mediate the acetylation of H3K27,have been shown to be involved in GSK3-mediated MLL leukemia stem cell transcriptional programs through their association with MEIS1.51Because CBP and P300 are fused to MLL in the MLL-CBP and MLLP300, which retain active histone acetyltransferase activity, the recruitment of BRD4 to H3K27-acetylated chromatin is crucial for MLL leukemia.Histone acetyltransferase activity is clearly required for the maintenance of MLL;this requirement suggests that the chemical inhibition of CBP or P300 presents a therapeutic strategy for treating MLL leukemia.Indeed, in 2015, the selective CBP/P300 inhibitor I-CBP112 has been reported to inhibit MLL-AF9 leukemia in vitro and in vivo.52In 2017,another potent and selective inhibitor,A-485,that showed inhibitory effects on MLL-AF9 leukemia cells was discovered.53
As mentioned earlier, MLL translocations generally occur on one allele of MLL gene to produce the MLL chimeras, whereas the other MLL allele could generate the wild-type MLL protein.Early studies involving shRNAs targeting the wild-type copy of MLL54and small molecules targeting the MLL methyltransferase activity by disrupting wild-type MLL-WDR5 interaction55have showed that wild-type MLL and methyltransferase activity are required for the maintenance of MLL leukemia.However, a recent study featuring the genetic perturbation of the MLL or MLL SET domain demonstrated that the wild-type MLL and MLL methyltransferase activity are dispensable for leukemia maintenance.56This finding is consistent with the existence of patient-derived MLL leukemia cell lines (such as ML2) with a deletion of the entire wild-type MLL locus.In addition, the methyltransferase activity of MLL is not necessary to maintain the expression of the direct target genes of MLL in hematopoietic populations or to facilitate MLL-AF9-mediated leukemogenesis,57thus suggesting that MLL methyltransferase activity is not an ideal target for MLL leukemia.Indeed, despite the similar expressions of the wild-type and chimeric MLL alleles at the mRNA level, we found that the wild-type MLL protein is much less abundant than the MLL chimeras due to rapid protein turnover in MLL leukemia cells.29
Menin (MEN1), as a common interacting protein between wild-type MLL and MLL chimeras,is an oncogenic cofactor for the initiation of MLL chimera-mediated leukemogenesis.58The interaction of MEN1 with the N-terminus of MLL is required for leukemic transformation,59and small molecules targeting the interaction show profound effects against MLL leukemia both in vitro and in vivo.60,61However,MEN1 is ubiquitously expressed and widely recognized as a tumor suppressor in endocrine organs such as parathyroid glands, pancreatic islet cells, and anterior pituitary gland.Whether targeting MEN1 would impair its function as a tumor suppressor in endocrine organs remains unclear.62
As the targets of MLL chimeras, cyclin-dependent kinase 6(CDK6) and Jumonji domain containing 1C (JMJD1C) were reported to be required for MLL leukemia.63,64Placke et al conducted an shRNA screening in MLL-AF9 AML cells and found that the CDK6 depletion or CDK4/6 inhibitor palbociclib preferentially inhibits leukemia cell proliferation and induces myeloid differentiation.65Similarly,JMJD1C has been identified from an epigenetic shRNA library screening and found to maintain the growth and colony formation of MLL-AF9 AML cells in vitro and in vivo.63Furthermore, JMJD1C has been demonstrated to substantially maintain AML stem cell frequency and block differentiation of MLL-AF9 leukemia through direct interaction with HOXA9 in a conditional mouse model.66As an RNA binding protein, Musashi2 (MSI2) maintains the MLL leukemia self-renewal program by directly interacting with and retaining the efficient translation of HOXA9,MYC,and IKZF2 mRNAs, while conditional deletion of Msi2 led to delayed leukemogenesis, reduced leukemia burden, and impairment of leukemia stem cell function in a murine MLL-AF9 leukemia model.67We recently showed that the histone chaperone complex chromatin assembly factor (CAF1) and its subunit CHAF1B are required for hematopoiesis, whereas CHAF1B overexpression promotes MLL by blocking myeloid differentiation transcription factors such as CEBPA.Abrogation of CHAF1B inhibits the progression of MLL-AF9 leukemia in vivo.68
5.TRANSCRIPTIONAL ADDICTION IN MLL LEUKEMIA
Since the discovery of the MLL gene and its translocation,numerous studies have attempted to uncover the molecular mechanism of underlying MLL pathogenesis despite the complexity of MLL fusion partners.Interestingly, an artificial MLL-βGAL fusion protein in a knock-in mouse model was found to result in the development of lymphoid and myeloid leukemias with reduced penetrance and long latencies.However, the β-galactosidase possesses neither inherent transcriptional activity nor any homology to MLL partner genes.β-Galactosidase can form a tetramer, thus suggesting that MLL- βGAL oligomerizes MLL to drive leukemia, albeit with a long latency period.69Further studies with dimerized MLL-FKBP,70MLL-AF1p, and MLL-GAS771showed that homo-oligomerization of MLL is necessary and sufficient for MLL leukemogenesis and activation of the downstream HOX genes and MEIS1.However, the molecular mechanisms of dimerized MLL chimeras in transcriptional regulation and MLL leukemogenesis remain unknown.72
The most common MLL translocation partners contain transcriptional activity and exist in two biochemically distinct complexes,namely,super elongation complex(SEC)and DOT1L complex, which cooperate to activate MLL target gene expression and provide insights into how MLL chimeras mediate the misregulation of transcription and leukemogenesis(Fig.1B).73In 2010, three groups74-76individually identified a large complex containing AF4/FMR2 (AFF) family protein(AFF1-4), the YEATS domain protein family members ENL or AF9, the Pol II elongation factors 11-19 lysine-rich leukemia(ELL)proteins,ELL-associated factor 1(EAF1)or EAF2,and PTEFb(composed of CDK9 and CCNT1).P-TEFb is essential for the transition from transcription pausing to elongation through the phosphorylation of the Pol II C-terminal domain(CTD)and negative elongation factor (NELF).77The DOT1L complex contains the subunits of DOT1L, which is a histone 3 lysine 79(H3K79) methyltransferase, AF10, and ENL or AF9.73,74,78Genetic perturbation of Dot1L in MLL-AF9 leukemia leads to decreased of H3K79 methylation, which may mediate the transcriptional elongation of MLL-AF9 targets.79,80However,although the DOT1L catalytic inhibitor EPZ004777 showed prolonged survival in a mouse MLL xenograft model,81phase I clinical trials suggest that the DOT1L inhibitors need to be used in combination with other therapies.29These data suggest that DOT1L complex may have catalytic-independent functions in MLL leukemia,and targeting the DOT1L protein itself via acute degradation appears to be a better option.Additionally,targeting of ENL in MLL-AF4 and MLL-AF9 leukemia cells showed that ENL is essential for MLL leukemia growth and that acute loss of ENL suppresses the initiation and elongation of RNA polymerase II at genes with high levels of ENL occupancy.82,83Taken together, these data demonstrate that SEC and DOT1L complexes are vital for the promotion of target genes expression in MLL leukemia through transcriptional regulation, thus highlighting a transcriptional addiction or dependency mechanism for MLL leukemia.
Transcriptional addiction is derived from oncogene addiction84that describes the behavior of cancer cells that depend on or are addicted to one or a few oncogenes for maintenance of phenotype and cell proliferation of cancer cells.These oncogenes are initially acquired during the multiple-step of oncogenesis and remain essential for maintaining the cancer cells long after they have become fully neoplastic.The transcription addiction extends the concept of oncogene addiction with other changes during tumorigenesis, including transcriptional dysregulation,enhancer malfunction, and epigenetic modulation.85Since MLL translocations are the driver mutations to promote MLL leukemogenesis and they are absolutely essential for MLL leukemia maintenance, targeting the transcriptional addiction mediated by MLL chimeras and their hijacked SEC and DOT1L complexes would be an appropriate therapeutic strategy for the aggressive MLL leukemia.
Within SEC complex, the AFF proteins serve as scaffolds for the binding of the other subunits and regulate multiple steps of transcriptional elongation (Fig.1).74-76P-TEFb is required for the phosphorylation of Pol II CTD and NELF to promote release from the promoter-proximal pausing, whereas ELL proteins could enhance the processivity of Pol II elongation.86-88Fusion of SEC subunits with MLL suggests that the oncogenic MLL chimeras activate MLL target gene expression through SEC recruitment to bypass normal transcription elongation checkpoints.20Moreover, impairment of the SEC function through shRNA-mediated depletion jeopardizes the MLL target gene HOXA9 in MLL leukemia, thus suggesting that SEC is an effective target for MLL leukemia treatment.Interestingly,MLLAF6, an MLL fusion with cytoplasmic protein that does not directly bind to the SEC or DOT1L complexes but contains a dimerization domain,constitutively recruits the SEC and DOT1L complexes to the target chromatin,which suggests that dimerized MLL fusion proteins activate transcription through the recruitment of these via unknown mechanisms.74
To prove the concept of targeting MLL chimeras and SEC in MLL leukemia, we analyzed the wild-type MLL and MLL chimeras in MLL leukemia and found that the wild-type form is much less abundant than the chimeras.Unbiased shRNA screening and mass spectrometry analysis revealed that the IRAK4-mediated pathways and E2/E3 hybrid ubiquitin-protein ligase UBE2O specifically control the protein degradation of wild-type MLL protein.29Targeting wild-type MLL degradation through IRAK4 inhibition or UBE2O depletion results in stabilization of the protein, which displaces the MLL chimeras and SEC from some of their target genes and leads to the impediment of MLL leukemia cell proliferation in vitro and in vivo.29Stabilization of MLL provides a new paradigm in the development of therapies for aggressive MLL leukemia by indirectly targeting MLL chimeras and SEC-mediated transcriptional addiction.89

Figure 2.Chronological summary of major findings and therapies targeting MLL leukemia.
To directly target SEC, we recently identified KL-1, a peptidomimetic lead compound, and its structural homolog,KL-2, through structure-based in silico screening and biochemical validation.86These two small molecules disrupt the interaction between AFF proteins and P-TEFb, leading to degradation of SEC complex in cells.Disruption of SEC impairs the release of Pol II from promoter-proximal pausing and reduces the transcription elongation rates for processive elongation.These two inhibitors are potent subjects in targeting multiple SEC-mediated biological processes and speculated to be promising compounds in targeting the transcriptional addiction mediated by MLL chimeras and SEC in MLL.90
6.SUMMARY
Despite significant advances in the understanding of the pathogenesis of MLL,no effective clinical treatment for this type of aggressive leukemia is yet available (Fig.2).Whether the mechanisms gained mostly from MLL-AF9, MLL-AF4, and MLL-AF4 could be generally applied to other MLL fusion proteins and MLL-PTD is unknown because of the diversity and complexity of the MLL recombinome.Moreover, no systematic comparison of the functions and mechanisms of these molecules in MLL leukemogenesis and maintenance has been published.The exact targets and transcriptional programs mediated by various MLL fusions remain unclear because different MLL fusions in different lineages have different target genes and transcriptional circuits.How the cell of origin, microenvironment,and types of MLL fusion affect the lineage specification of MLL remains undetermined.
Although numerous cooperating events,including MLL target genes, downstream and parallel pathways, have been identified for MLL leukemia based on the two-hit hypothesis,they are not necessarily useful in translation if targeting of these events impairs the normal hematopoiesis.Why MLL relies on these cooperating events more than normal hematopoiesis is unclear.Moreover, because these events are cooperative and not the drivers of mutations for MLL, completely blocking the progression of MLL by targeting these events is insufficient; in fact, resistance may be expected to arise shortly.49,50Such complexities are the major challenges impeding the development of effective therapeutic strategies for MLL leukemia.Thus far,none of these target therapies or drugs has been approved to specifically target the aggressive MLL leukemia.Targeting the transcriptional addiction mediated by MLL chimeras and their associated SEC and DOT1L complexes seems to be a better option for obtaining effective MLL therapies.Understanding the mechanisms underlying SEC and DOT1L complex recruitment to chromatin by MLL fusions, how these molecules promote MLL leukemogenesis through their target genes, and how they cooperate to drive transcriptional misregulation of the MLL target genes are important to reveal the transcriptional addiction in MLL leukemia and develop suitable treatment approaches.Thus, targeting the transcriptional addiction in MLL leukemia may serve as a novel paradigm for the target therapy of various cancers.
ACKNOWLEDGMENTS
We apologize for the work that could not be recognized here due to space constraints.We thank Edwin R.Smith for discussing and sharing his views.This work was supported by grant from the “Thousand Young Talent Program” awarded to K.L.
杂志排行

血液科学的其它文章
- Successful ex vivo expansion of mouse hematopoietic stem cells
- A chemotaxis model to explain WHIM neutrophil accumulation in the bone marrow of WHIM mouse model
- Interleukin-12 supports in vitro self-renewal of long-term hematopoietic stem cells
- The disruption of hematopoiesis in tumor progression
- Progress of cGVHD pathogenesis from the perspective of B cells
- Molecular mechanisms for stemness maintenance of acute myeloid leukemia stem cells