高效液相色谱法同时测定灵芝糖浆中4种成分*
2019-11-01黄强燕张佳莉王倬顾峥嵘
薛 平,黄强燕,李 莉,承 晨,张佳莉,王倬,姚 晓,顾峥嵘
(江苏省常州市食品药品监督检验中心,江苏 常州 213000)
灵芝糖浆是由灵芝制成的单方制剂,功能养心安神、健脾和胃,用于心悸、失眠、食欲不振及神经衰弱。其制备工艺主要是将灵芝粉碎成粗粉,照流浸膏剂与浸膏剂项下的渗漉法,用80%乙醇作溶剂,浸渍1周后,缓慢渗漉至无色,收集渗漉液,回收乙醇;药渣加水煎煮2次,每次2 h,合并煎液,滤过,滤液与渗漉液合并,浓缩至相对密度为 1.16~1.18(60 ℃),加入蔗糖 600 g及苯甲酸钠适量溶解后,滤过,加水调整总量至1 000 mL,搅匀,即得[1-3]。灵芝含有多糖类、氨基酸、多肽、三萜类、生物碱等多种化学成分,具有镇静安神、降血糖、保肝解毒、抗肿瘤、免疫调节等作用,其中三萜类和多糖类化学物质被认为是主要有效成分,常作为衡量灵芝质量的依据[3-5]。目前灵芝糖浆质量控制仍执行《中华人民共和国卫生部药品标准·中药成方制剂(第十七册)》,无指标性成分控制。本研究中从三萜中的灵芝酸类成分角度出发,采用高效液相色谱(HPLC)法测定了市场上常见厂家不同批次的产品,为该制剂的质量控制提供参考。现报道如下。
1 仪器与试药
1.1 仪器
Thermo U3000型高效液相色谱仪(美国赛默飞世尔科技公司),包括 LPG-3400SDN泵,WPS-3000型TLSL Analytical自动进样器,TTC-3000RS型柱温箱,DAD-3000型二极管阵列检测器;XP105DR型电子天平(十万分之一,瑞士梅特勒-托利多公司)。
1.2 试药
灵芝酸 C2(批号为 180517,含量为 98.19% ),灵芝酸 G(批号为 18032609,含量为 99.30%),灵芝酸 A(批号为18032603,含量为98.26%)均购于成都普菲德生物科技有限公司;灵芝酸B(北京世纪奥科生物技术有限公司,批号为171030,含量为98.15%)。水为超纯水,乙腈、甲醇为色谱纯,其他试剂均为分析纯。
灵芝糖浆分别来自5个不同厂家,即江苏新先制药有限公司、江西省芙蓉堂药业股份有限公司、大理白族自治州中药制药有限公司、株洲千金药业股份有限公司和江西杏林白马药业有限公司,编号分别为A,B,C,D,E。详见表 1。
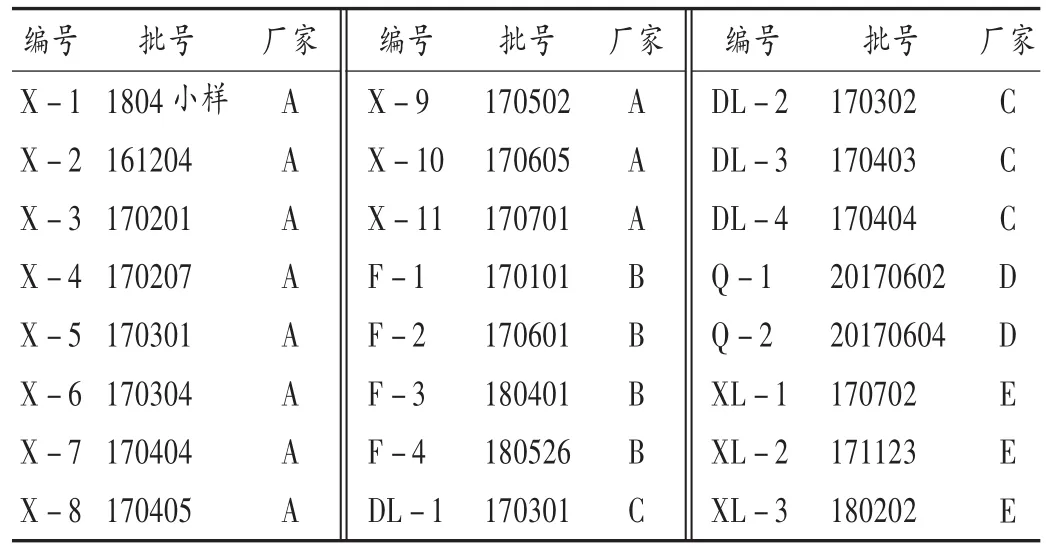
表1 样品信息
2 方法与结果
2.1 色谱条件[6-13]与系统适用性试验
色谱柱:安捷伦Zorbax Eclipse Plus C18柱(250 mm×4.6 mm,5 μm);流速:0.8 mL /min;流动相:乙腈 -0.05 moL 磷酸二氢铵水溶液(27 ∶73,V/V),等度洗脱;柱温:15 ℃;检测波长:254 nm;进样量:10 μL。在此色谱条件下的色谱图见图1。取线性关系考察项下同一浓度的混合对照品溶液,按拟订色谱条件进样,记录色谱图,灵芝酸C2、灵芝酸G、灵芝酸B、灵芝酸A峰的理论板数均不得低于3 500,分离度均大于1.5。

图1 高效液相色谱图
2.2 溶液制备
分别取灵芝酸C2、灵芝酸G、灵芝酸B、灵芝酸A对照品适量,精密称定,置100 mL容量瓶,加甲醇溶解并稀释至刻度,制成每 1 mL含灵芝酸 C2100.1 μg、灵芝酸 G 100.3 μg、灵芝酸 B 100.4 μg、灵芝酸 A 100.2 μg的混合对照品贮备液。精密量取25 mL样品溶液,用乙酸乙酯振摇提取5次,每次25 mL,合并乙酸乙酯提取液,乙酸乙酯提取液回收溶剂至干,残渣加甲醇溶解并转移至10 mL容量瓶中,加甲醇至刻度,摇匀,用0.45 μm微孔滤膜过滤,取续滤液,即得供试品溶液。取空白试剂(水),按供试品溶液制备方法制备阴性对照品溶液。
2.3 方法学考察
专属性试验:分别取 2.2项下 3种溶液,进样10 μL测定。阴性对照品溶液色谱中,在与对照品溶液及供试品溶液色谱峰相应处无4种被测组分的峰出现,阴性对照无干扰,专属性良好(图1)。
线性关系考察:分别精密量取2.2项下混合对照品贮备液,精密吸取1,2,5 mL,分别置100 mL容量瓶;量取5 mL,置50 mL容量瓶,量取5 mL和8 mL分别置10 mL容量瓶。加入甲醇稀释至刻度,制成6个不同质量浓度的对照品溶液,按拟订色谱条件进行测定,进样10 μL,以峰面积值(Y)为纵坐标、质量浓度(X,μg/mL)为横坐标进行线性回归。结果见表2。

表2 4种成分的线性回归方程和线性范围
精密度试验:精密吸取线性关系考察项下同一浓度的混合对照品溶液,按拟订色谱条件重复进测定6次。结果灵芝酸C2、灵芝酸G、灵芝酸B和灵芝酸A的峰面积RSD分别为 0.48%,0.80%,0.78%和0.42%(n=6),表明仪器精密度良好。
重复性试验:精密量取样品(编号X-1)25 mL,依法制备6份供试品溶液,按拟订色谱条件测定。结果灵芝酸C2、灵芝酸G、灵芝酸B和灵芝酸A峰面积的RSD分别为 1.70%,1.35%,0.73%,1.29% (n=6),表明方法重复性良好。
稳定性试验:精密量取样品(编号X-1)25 mL,依法制备供试品溶液,分别在 0,4,8,12,16,24 h 时测定。结果灵芝酸C2、灵芝酸G、灵芝酸B和灵芝酸A峰面积的RSD分别为 1.59%,0.57%,0.73%,0.50% (n=6),表明供试品溶液在24 h内稳定。
加样回收试验:精密量取样品15 mL(编号X-1),分别加入灵芝酸 C2(45.20 μg /mL)1 mL,灵芝酸 G(102.7 μg /mL)1 mL、灵芝酸 B(40.50 μg /mL)0.5 mL和灵芝酸 A(81.10 μg/mL)1 mL,并稀释至 25mL,依法制备供试品溶液,并测定含量,计算回收率。结果见表3。

表3 加样回收试验结果(n=6)
2.4 样品含量测定
精密量取不同厂家各批次灵芝糖浆样品,依法制备供试品溶液,并测定含量。结果见表4。
3 讨论
3.1 色谱条件优化
前期试验参考文献[6],使用流动相为乙腈-0.04%甲酸溶液梯度洗脱相同的色谱柱,经过调整后未能使目标峰达到较好地分离,曾考察流动相乙腈与0.05 moL/L磷酸二氢铵水溶液[9]或各种酸(0.1% 磷酸、0.1% 甲酸和 0.1% 冰醋酸)[6-13]、流动相流速(0.8 mL /min 和1.0 mL /min)、柱温(15,25,35,40 ℃)[9-12],发现流速为0.8 mL /min、流动相为乙腈 -0.05 moL /L 磷酸二氢铵水溶液(27 ∶73,V/V)、柱温为 15 ℃时,目标峰峰形较好,且分离效果较好,纯度较高。检测波长参考相关文献并结合实际情况,发现在254 nm波长处目标峰有较大吸收[6-13],适宜作为含量测定的检测波长。曾考察不同品牌色谱柱[250 mm × 4.6 mm,5 μm 的色谱柱(安捷伦Zorbax Eclipse PLus C18色谱柱、安捷伦Zorbax SB-C18色谱柱、岛津InertsiL ODS-3液相色谱柱和沃特世Waters X Bridge色谱柱)]均能使目标峰达到较好的分离效果和纯度。
3.2 提取条件优化
前期试验发现,4种灵芝酸含量相对较高,故选取为测定指标性成分。由于样品中4种灵芝酸含量普遍较低,且含有大量的蔗糖等水溶性物质,所以测定前需要提取纯化,并浓缩样品。供试品溶液制备过程前期,考察了不同的提取溶剂[13-16](石油醚 30 ~60 ℃ 、石油醚60~90℃、乙醚、二氯甲烷、丙酮、乙酸乙酯和甲酸乙酯)、提取过程加不加酸(甲酸)、最佳溶剂的提取次数(2,3,4,5,6 次)。根据试验结果、提取效率和使用便利性,发现无需加酸,采用乙酸乙酯萃取5次,就可达到较满意的提取效果。
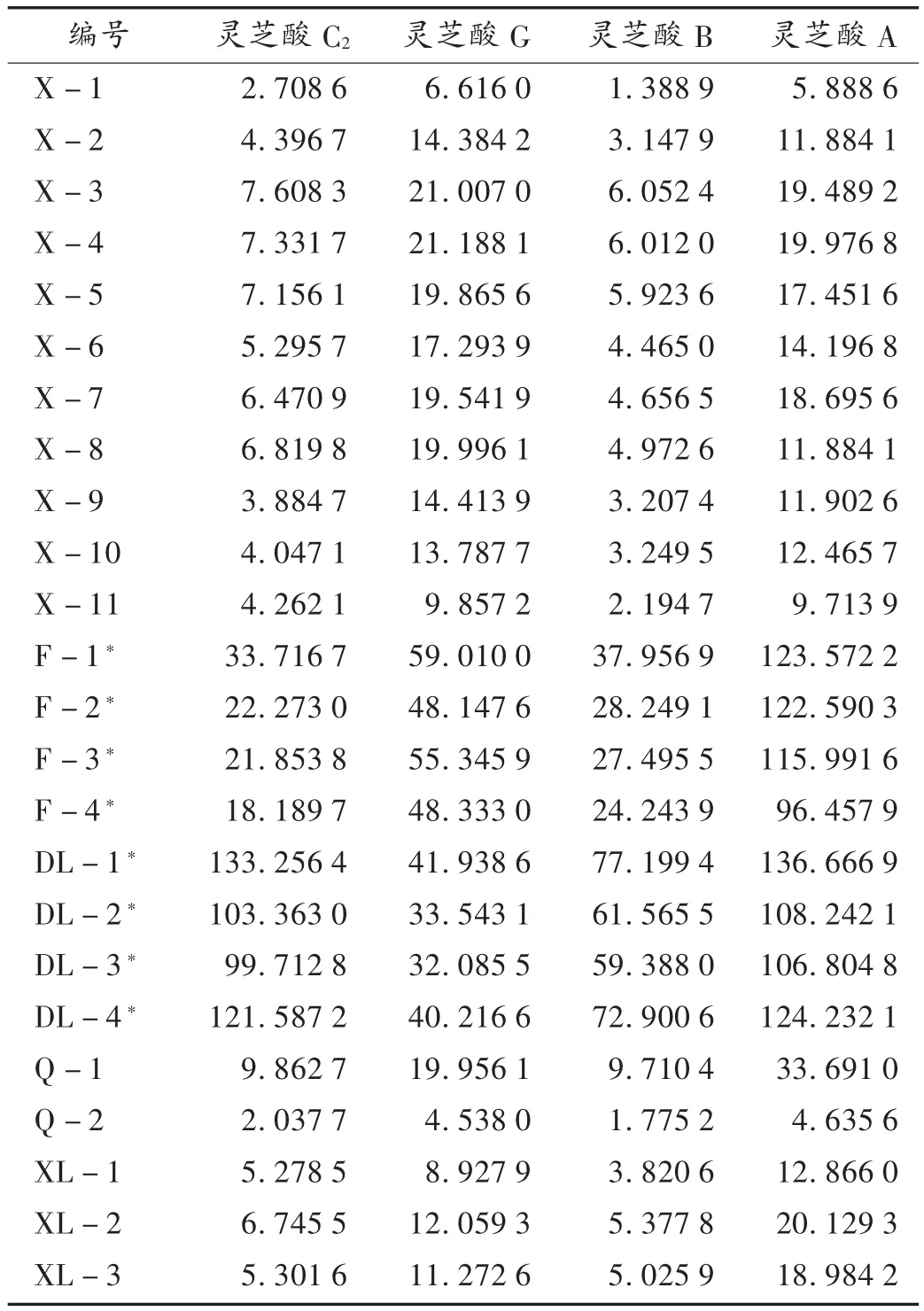
表 4 24批样品含量测定结果(μg/mL,n=3)
3.3 结果分析
试验过程中发现,不同厂家的样品含量具有显著差异,生产厂家B(灵芝酸A)和生产厂家C(灵芝酸C2、灵芝酸A)2家的样品部分灵芝酸已超出曲线范围,推测可能由于原料和制剂工艺的差异性导致不同品牌之间含量差异显著。测定了生产厂家A一批灵芝原料、样品中间体,与厂家面对面交流后发现,在生产灵芝糖浆过程中,醇提渗漉液回收乙醇后,会出现大量的沉淀,为了后期样品处理方便和得到最终完美清澈的溶液型样品,弃去了大部分沉淀(脂溶性成分),对此部分沉淀的处理工艺控制不严,导致大量的灵芝酸类成分被弃去。这可能是同一品牌不同批次产品不稳定,不同品牌灵芝糖浆灵芝酸类含量差异较大的主要原因。
目前,该制剂标准仅检测了相对密度,缺乏专属性检查,导致市面上缺乏对该制剂品质的衡量标尺,4种灵芝酸含量相对较高,具有代表意义,方法相对简便,结果可靠,可为厂家灵芝糖浆质量控制与品质评价提供参考。
