钠离子电池正极材料β-NaMnO2的容量衰减机制
2019-10-31刘晨子胡业旻李文献胡鹏飞金红明朱明原
刘晨子,胡业旻,李文献,刘 杨,胡鹏飞,金红明,朱明原,李 瑛
(上海大学材料科学与工程学院,上海200444)
近年来,电动汽车、混合动力汽车及分布式能源储存系统快速崛起,使锂离子电池的需求量剧增.然而锂储量有限,使得锂离子电池价格不断攀升,这将制约锂离子电池的大规模应用[1-4].钠和锂在元素周期表中属于同一主族,具有相似的物理化学性质.相比于锂,钠的储量十分丰富,且分布广泛、提炼简单,因此在规模化储能应用方面,钠离子电池更具优势,近年来对钠离子电池的研究也越来越多[5-8].与锂离子相比,钠离子半径较大,使得钠离子在充放电过程中需要阴阳极材料具有更大的嵌入空间,故电极材料的选择至关重要.只有研制出具有稳定脱嵌钠能力的电极材料,才能实现钠离子电池的实用性突破[9].这里,高稳定性的正极材料是目前钠离子电池发展的一大瓶颈[10],在选择正极材料时同时要兼顾环境与成本因素.锰氧化物资源丰富、成本低廉、毒性低,且具有多种隧道或层状结构,是极具潜力的钠离子电池正极材料体系,因此NaxMnO2(0.2≤x≤1)成为当前研究热点之一[11-13].但是,钠离子电池存在循环寿命短、容量衰减快等问题[13],NaxMnO2类电极材料也不例外[14-16].
β-NaMnO2材料具有特殊的之字形层错结构,且钠离子传输通道良好、制备简单、理论容量高,是一种很有应用前景的钠离子电池正极材料.Abakumov等[17]等报道了2种NaxMnO2类材料,一种是没有孪晶面的α-NaMnO2,而另一种是每一个NaMnO2层上都具有孪晶面的β-NaMnO2,但是并未进行电化学性能的研究;Cl´ement等[18]等建立了β-NaMnO2的晶体结构模型,并模拟了充放电过程中晶体结构的变化;Billaud等[19]等发现,在脱嵌钠的过程中会发生一定的堆垛层错,同时伴随着结构无序化的现象.以上研究中均出现了前几次循环容量衰减较快的现象,但是目前关于该机制的研究并未很透彻.本工作通过简单固相反应制备了层状β-NaMnO2,并采用X射线衍射(X-ray diffraction,XRD)、扫描电子显微镜(scanning electron microscopy,SEM)、高分辨透射电子显微镜(high resolution transmission electron microscopy,HRTEM)、X射线光电子能谱仪(X-ray photoelectron spectroscope,XPS)等手段对充放电过程中材料结构和形貌进行表征,以探究容量衰减原因.
1 实 验
1.1 β-NaMnO2材料的制备、电池装配及电化学性能测试
按照质量比1.05∶1称量Na2CO3(AR)和Mn2O3(AR).以10∶1的球料比,将称好的原料放入转速为300 r/min的球磨机中,球磨10 h后取出压片;然后放入管式炉中,以5°C/min的速度升至950°C,保温10 h后取出,即得到层状β-NaMnO2;最后迅速将样品转移进手套箱冷却至室温.
按8∶1∶1的质量比例称量160 mg的β-NaMnO2、20 mg的导电炭黑和20 mg的聚偏氟乙烯(polyvinylidene fluoride,PVDF),加入适量N-甲基吡咯烷酮(N-methyl pyrrolidone,NMP)溶剂进行混合.混合物浆料用磁力搅拌器搅拌24 h,将搅拌均匀的浆料以150 μm的厚度均匀涂抹在铝箔表面,在120°C下真空干燥12 h,使溶剂挥发;然后取出极片,裁成直径为15 mm的小圆片.负极采用纯度为99.9%的金属钠片,片厚约为0.4 mm,直径约为10 mm.用1 mol/L的无水NaClO4(AR)溶于碳酸丙烯酯(propylene carbonate,PC,AR)作为电解液,玻璃纤维滤纸为隔膜,在充满氩气的手套箱(米开罗那Super 1220/1750)中装配成CR2032型扣式电池.采用Land CT2001A电池性能测试系统(武汉金诺电子有限公司)进行充放电测试,电压范围为1.5~4.0 V,电流密度分别为10,20和50 mA/g.在室温下采用CHI1030b型电化学工作站(上海辰华)进行循环伏安性能测试,测试电压范围为1.5~4.0 V,电压扫描速度为0.1 mV/s.
1.2 电池的拆解及正极物质的非原位表征
将前3次恒流充放电循环中分别处于充电态和放电态的电池从Land测试仪上取下,放置于手套箱中;利用钳子等工具将电池拆封,取出正极片在碳酸丙烯酯溶液中清洗几次;然后在手套箱中自然晾干.非原位XRD直接用该正极片进行测试;将正极片剪成面积约为0.25 cm2的小片进行非原位XPS测试;非原位TEM测试则通过刮下部分极片上的正极材料,用丙酮超声稀释后,滴在透射微栅上进行.
1.3 材料的表征
采用Rigaku公司生产的D/MAX-R2C型XRD仪对该材料进行物相分析,测试条件为:Cu靶,管电压为40 kV,管电流为100 mA,连续扫描速度分别为4°/min和1°/min,步宽为0.02°.本工作采用JEOL(日本电子)公司的JSM-7500F型冷场发射SEM进行形貌表征,工作电压为15 kV.采用JEOL(日本电子)公司生产的JEM 2100F型高分辨分析型场发射TEM进行微区结构分析,用能量色散谱(energy dispersive spectroscopy,EDS)仪辅助分析样品成分,加速电压为200 kV.采用英国Thermo Fisher公司ESCALAB 250Xi型的XPS进行样品表面元素价态分析.
2 结果与讨论
2.1 电化学性能
图1(a)为β-NaMnO2作为钠离子电池正极材料在10 mA/g的电流密度下第1、2和5次充放电曲线,电压范围为1.5~4.0V.该材料的首次放电比容量高达184mA·h/g,这与Billaud等[19]报道的结果相近.但是第2次循环的放电比容量迅速衰减至140 mA·h/g左右,并且随着循环次数的增加容量持续衰减,第5次降至111 mA·h/g.图1(b)分别为第一次充电和放电结束后样品的EDS测试结果.可以看出,充电结束后大部分钠迁出正极活性材料,微米棒上残留的钠很少;而放电过程结束后,钠又回到了微米棒中来,这和常规充放电过程中钠的变化规律是一致的.图1(c)为β-NaMnO2在10 mA/g的电流密度下的循环性能图.经过10次循环,容量剩余104 mA·h/g,保持率为73%,衰减较多;同时随着充放电次数的增加,材料的库伦效率从80%增加到90%,且趋于稳定.图1(d)为样品在不同电流密度下的放电比容量.由图可知,在10 mA/g电流密度下,放电比容量由142 mA·h/g降至109 mA·h/g,衰减较大;随着电流密度的不断增大,放电比容量逐渐减小;在100 mA/g时,放电比容量仅为37 mA·h/g;当电流密度再次回到10 mA/g时,容量恢复至60 mA·h/g左右,容量保持率仅为43%.
2.2 结构与形貌
为了探究β-NaMnO2容量衰减的原因,对经过前3次充放电循环的电极材料进行XRD测试,分析材料的物相变化,并与未进行充放电测试的β-NaMnO2进行比较(见图2).图2下方为所制备β-NaMnO2样品的XRD图谱,样品的所有衍射峰均与β-NaMnO2(PDF#72-0412)的特征衍射峰符合得较好,峰形尖锐,不存在明显的杂质峰.从图2上方经过3次充放电后样品的XRD图谱可知,前3次充放电之后,衍射峰的变化趋势基本一致.充电结束后,分别在12°,25°左右出现较明显的Na0.91MnO2的特征峰,在29°左右出现Na0.7MnO2的特征峰,且Na0.91MnO2的特征峰强于Na0.7MnO2,表明在充电后Na0.91MnO2的结晶度比Na0.7MnO2好.但是,25°左右的Na0.91MnO2的特征峰较宽,说明钠离子从MnO6八面体中脱出后,该结构稳定性降低[19],这可能是因为随着充电反应的进行,锰的价态发生了变化,Mn3+被氧化成了Mn4+.同时,由于NaxMnO2中钠含量的降低,使结构产生了Jahn-Teller缺陷,引起c轴方向上Mn-O层的变形或相对移动[20].

图1 β-NaMnO2的电化学性能Fig.1 Electrochemical profiles of β-NaMnO2

图2 β-NaMnO2的XRD图谱和10 mA/g电流密度下,样品第1至第3次充放电的XRD图谱(1.5~4.0 V)Fig.2 XRD pattern of the as-prepared β-NaMnO2and XRD patterns of β-NaMnO2 recorded during the first three electrochemical cycles at 10 mA/g between 1.5 and 4.0 V
当放电过程结束后,Na0.91MnO2和Na0.7MnO2的特征峰依然存在,而且Na0.91MnO2的特征峰变得尖锐,说明部分钠离子重新嵌入到MnO6八面体中,使结构变得稳定,因此放电后Na0.91MnO2的结晶度高于充电后的.而当充放电循环结束后,XRD图谱上并未出现β-NaMnO2的特征峰,这可能是由于随着钠离子在NaMnO2材料中的嵌入脱出,晶体结构发生了一定程度的变形,使钠离子重新嵌入受到阻碍,部分Mn4+不能被重新还原为Mn3+;而Mn4+与氧的结合更加牢固[21],导致NaMnO2发生相变,转变成了Na0.91MnO2和Na0.7由于完成循环后活性材料是涂覆在铝箔上直接进行XRD测试,因此XRD图谱上出现Al峰属正常现象;另外,XRD图谱上会出现一些未知峰,有待进一步研究.

图3 β-NaMnO2的电子显微镜照片Fig.3 Electron microscopic images of β-NaMnO2
图3 (a)是所制备β-NaMnO2的SEM图,(b)是(a)的TEM图.可见所制备β-NaMnO2呈微米棒状,平均长度约为30 μm,宽度约4 μm,棒的表面有一些颗粒状附着物,可能是在高温烧结过程中过量碳酸钠分解后残留的氧化钠[18].由(b)可见,β-NaMnO2的晶格条纹清晰,且沿着棒长径方向生长.对β-NaMnO2进行标定,测出晶面间距d1=0.21 nm,d2=0.24 nm,分别对应其(012)和(110)晶面方向.为了进一步分析样品在充放电过程中的微结构变化,分别对经过充放电循环的电极材料进行HRTEM表征(见图3(c)和(d)).从(c)中可以看出,第一次充电结束后,随着钠离子的脱出,微米棒由许多不同取向的微小纳米晶组成.标定结果显示,其中既有Na0.91MnO2的相存在,也有Na0.7MnO2的相存在,如图中红线区域所示.从上述情况可以看出,第一次充电结束后发生了由NaMnO2到Na0.91MnO2和Na0.7MnO2的相转变.除了相转变,在充电完成的HRTEM图上还发现了一些结构缺陷的存在,如肖特基(Schottky)点缺陷和堆垛层错缺陷[23],如(c)中白色方框区域所示.这些缺陷和多晶区域对应图2中XRD图谱上较宽广、强度不高的衍射峰.图3(d)和(e)是第一次放电结束后样品不同区域的HRTEM图.从图3(d)中可以看出,有2个方向的晶格条纹出现,经标定后发现,其晶面间距分别为d1=0.27 nm,d2=0.20 nm,分别对应Na0.91MnO2的(200)晶面和(120)晶面.图3(e)中标定出了0.28 nm的晶面间距,经比对该晶面属于Na0.7MnO2的(004)晶面,这与图2中XRD图谱所示的样品相变规律是一致的.
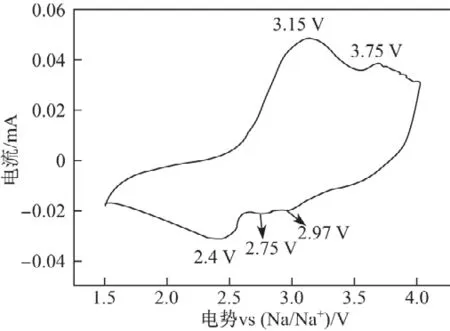
图4所制备样品的循环伏安曲线(1.5~4.0 V,0.1 mV/s)Fig.4 CV curve of the as-prepared sample(1.5~4.0 V,0.1 mV/s)
图4 是样品经过几个充放电循环,性能稳定后的循环伏安曲线.图中有2个较平滑的氧化峰在3.15和3.75 V出现,这是因为此处发生了缓慢的氧化还原反应,其中3.15 V的氧化峰对应Mn3+被氧化为Mn4+的过程[24],而3.75 V的氧化峰可能是电解液同电极材料发生了副反应[25].在2.40,2.75和2.97 V处有3个还原峰出现,其中2.40 V处的还原峰对应Mn4+→Mn3+的还原反应;而在2.75和2.97 V出现的还原峰不明显,说明钠的嵌入反应可能是一个复杂的多相反应[26],在放电过程中只有部分Mn4+被重新还原成Mn3+,剩余少量Mn4+化合物则与电解液发生反应[27].
图5为所制备β-NaMnO2、第一次充电结束和第一次放电结束后电极材料的Mn 2p峰拟合谱图.对图5(a)中Mn 2p的峰进行拟合之后发现,652.8 eV和642.1 eV处谱峰分别为Mn3+2p 1/2和Mn3+2p 3/2的峰,说明制得的样品中Mn的价态为+3价,进一步表明制得的样品为纯净的β-NaMnO2.第一次充电结束后的Mn 2p的XPS图谱拟合结果如图5(b)所示,图中原始谱图上出现2个明显的峰.对这2个峰进行分峰拟合发现,Mn 2p 3/2峰的3个拟合峰分别出现在641.6,643.4和646.9 eV;相应地,Mn 2p 1/2的峰可以分为652.8,654.0和656.1 eV,分别对应着Mn4+,Mn3+和Mn7+.经过计算,Mn4+和Mn3+峰的总面积比例约为2∶1,这说明在充电结束后大部分Mn3+被氧化为Mn4+[28].但是,在充电反应过程中,646.9和656.1 eV位置处均有Mn7+的峰出现,这可能是由于电解液中的NaClO4使Mn4+发生了过度氧化[29].同时从(a)也可以看出,Mn7+的含量很少,因此在XRD精度条件有限的情况下,并不能检测到Mn7+相的存在.图5(b)是第一次放电结束后的Mn 2p峰拟合谱图,从图中可以看到646.9和656.1 eV位置处的峰几乎消失,也就是说在放电反应结束后,Mn7+重新被还原为低价态的锰.与图5(a)不同的是,Mn4+和Mn3+峰的总面积比例发生了变化,比例约为1∶2,这说明在钠离子重新嵌入结构的过程中,大部分Mn4+又重新被还原成了Mn3+,这与第一次充放电结束后的XRD结果是一致的,即Na0.91MnO2和Na0.7MnO2的峰在放电后变尖锐的同时又变强.除了利用分峰拟合方式对元素价态进行分析之外,ΔBEMn-O(Mn-O构型的Mn 2p 3/2和O 1s峰之间的结合能差)也被认为是评判不同Mn化学状态的标准[30].该差值的高低决定着不同价态的锰与氧结合能力的强弱.图6分别为第一次充电和第一次放电结束后的Mn-O结合能差值ΔBEMn-O.由图可知,第一次充电结束后ΔBEMn-O为111.5 eV,而第一次放电后ΔBEMn-O为111.1 eV,比充电态略低,说明充电态的Mn-O结合能较大.对于同一种元素,价态越高,与氧的结合力通常就越强[31],也就是说Mn4+在充电态时占的比例较大,这与图5分峰拟合的结果相一致,同时也验证了图2的XRD的结果.
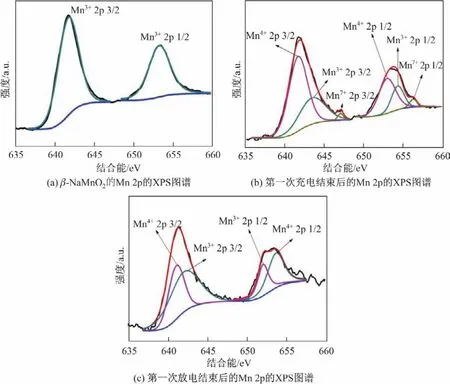
图5 β-NaMnO2的XPS图谱Fig.5 XPS spectra of β-NaMnO2
3 结束语
本工作通过简单固相反应制备了β-NaMnO2,首次放电比容量为184 mA·h/g,但衰减较快,10次充放电循环之后仅有73%的容量剩余.XRD测试结果表明,在充放电过程中产生Na0.91MnO2和Na0.7MnO2的新相,而β-NaMnO2相消失不见,这可能是因为充电时Mn3+氧化为Mn4+的过程中,发生了Mn-O层沿c轴方向的拉伸.由于发生了结构变形,钠离子脱出之后原来的晶体结构被破坏,使得钠离子难以在放电时重新嵌入,导致接下来的循环中β-NaMnO2相不再出现.充电后HRTEM图像上也出现了许多不同取向的纳米晶,甚至出现点缺陷和面缺陷,说明结构遭到了破坏.XPS测试结果显示,充电时大部分Mn3+被氧化为Mn4+,同时伴有Mn7+的出现;放电时Mn7+消失,大部分Mn4+转变为Mn3+.根据以上结果可以推断,β-NaMnO2材料容量衰减较快的机制是由于充电过程中的相转变造成结构变形,不利于钠离子在放电时重新嵌入,因而不能在放电时恢复β-NaMnO2相.该发现有助于进一步研究β-NaMnO2的电化学反应机制,对探索性能稳定的钠离子电池正极材料是有益的.

图6 Mn 2p和O 1s峰的高分辨XPS图谱Fig.6 High-resolution XPS spectra of Mn 2p and O 1s
