在疫苗生产和医疗行业中不同物品同载进行脉动真空灭菌的效果验证
2019-10-30王舰兵金鑫张可明
王舰兵 金鑫 张可明
[摘要]目的 驗证在疫苗生产和医疗行业中不同种物品同载进行脉动真空灭菌的效果。方法 用专业温度探头、生物指示剂通过化学和物理方法对同载的不同物品进行脉动真空灭菌。首先进行脉动真空灭菌器空载保压试验和布维-狄克(B-D)试验及热穿透试验,确认灭菌器性能合格后将疫苗生产中常用的硅胶管道、敷料包、橡胶栓、过滤器、超声破碎仪、玻璃吸管、无菌服、止血钳及器械等同芽孢指示剂一起按照规定的点位放入灭菌器内进行132℃、8 min挑战试验,共3次。最后进行生物挑战试验和无菌试验。结果 脉动真空灭菌器保压程序停止900 s后,初始压力仍≤-93 kPa,泄露率≤2 kPa,保压试验合格;在空载条件下脉动上限+75 kPa、脉动下限-75 kPa、灭菌温度132℃、灭菌时间4 min,完成灭菌操作后,取出测试包并打开观察B-D指示图呈现色列条变化均匀与对照图设计的颜色一致,灭菌器真空性能试验合格;在灭菌过程中的任何相同时间不同位置间的温度差别(均一性)均≤2℃,且灭菌器温度控制探头与监控记录仪记录的温度差值均≤1℃,空载热分布试验合格;按着规定的装载方式且达到满负荷装载后选择132℃、8 min、脉动±75 kPa灭菌程序进行试验,所有的测试各点标准灭菌时间(F0)≥15 min,满载热穿透试验合格;被灭菌后的生物指示剂管内颜色呈现紫色,与说明书判定合格颜色一致,为培养阴性(-),同时55~56℃培养24 h后无细菌生长,微生物挑战试验合格;物品的淋洗水在接种培养基中放置在35℃培养箱中培养3 h后,培养基中没有菌落生长,无菌试验合格。结论 疫苗生产和医疗行业中常用的多种不同物品可以用脉动真空灭菌器同时进行132℃、8 min灭菌,且能达到国家药典规范的要求。
[关键词]脉动真空;布维-狄克试验;验证效果;微生物挑战试验;无菌保证水平
[中图分类号] R953 [文献标识码] A [文章编号] 1674-4721(2019)8(c)-0172-04
[Abstract] Objective To verify the effect of pulsating vacuum sterilization of different species in the vaccine manufacturing and medical industries. Methods The pulsating vacuum sterilization of different articles on the same load were carried out by chemical and physical methods with professional temperature probe and biological indicator. Firstly, no-load pressure holding test and B-D test of pulsating vacuum sterilizer and thermal penetration test were carried out to confirm that the performance of sterilizer was qualified, and then the silica gel pipes, dressing bags, rubber bolts, filters, ultrasonic crushers, glass pipettes, aseptic clothes, hemostatic forceps and other spore indicators commonly used in vaccine production were identified, challenge tests at 132℃ and 8 min were carried out in the sterilizer at the prescribed locations for three times. Finally, microbial challenge test and aseptic test were carried out. Results The initial pressure of pulsating vacuum sterilizer was still ≤-93 kPa and the leakage rate was ≤2 kPa after 900 seconds of the pressure-holding procedure of pulsating vacuum sterilizer, the pressure-holding test was qualified. Under no-load condition, the upper limit of pulsation +75 kPa, the lower limit of pulsation -75 kPa, sterilization temperature 132℃, sterilization time 4 min. After sterilization operation, the test kit was taken out and the B-D indicator chart was opened to observe that the change of Israel strip was uniform and the color of the control chart was the same, the vacuum performance test of the sterilizer was qualified. The temperature difference (homogeneity) between different locations at the same time during sterilization was less than 2℃, and the temperature difference recorded by the sterilizer temperature control probe and the monitoring recorder was less than 1℃, and the no-load heat distribution test was qualified. According to the prescribed loading mode and after full load loading, the sterilization procedure of 132℃, 8 min and pulsation±75 kPa was selected to carry out the test. The standard sterilization time (F0)≥15 min at all test points, and the full load heat penetration test was qualified. After sterilization, the color in the tube of the bio-indicator was purple, which was consistent with the qualified color determined by the instructions. It was negative for culture (-). At the same time, there was no bacterial growth after incubation at 55-56℃ (-) for 24 hours, and the microbial challenge test was qualified. The leaching water of the product was placed in the incubator at 35℃ (-) for 3 hours, but not in the culture medium. Colony growth, sterility test was qualified. Conclusion Various kinds of different articles commonly used in vaccine production and medical industry can be sterilized simultaneously by pulsating vacuum sterilizer at 132℃ for 8 minutes, and can meet the requirements of the national pharmacopoeia.
[Key words] Pulsating vacuum; Bowie-Dick test; Verification effect; Microbial challenge test; Sterility assurance level
高压灭菌是使物品灭菌后达到最终无菌的要求,是药品生产及相关行业中的一个基本保证[1],是控制染菌的有效措施之一[2]。被灭菌的物料及器械由于材质、包装、体积、耐热系数等各不相同,同载灭菌时在相同的灭菌温度和灭菌压力下是否出现灭菌不彻底的现象,给生产和医疗带来染菌的后果。为了保证高压蒸汽灭菌物品的灭菌质量[3],根据被灭菌物品的特性采用—种或多种方法组合灭菌。灭菌后还要采取适当方法进行灭菌效果的验证[4],高压灭菌效果验证是最关键的步骤,同时在药品生产、医疗行业发展过程中,用验证的方法是每个国家检验质量管理体系稳定运行和发展的重要手段[5],同时也是质量管理规范(GMP)不断发展创新的可靠保证。验证是更加安全和更加先进的质量保证方法[6]。现在多数制药行业已放弃了老式的下排汽高压蒸汽灭菌器,采用了安全可靠、灭菌速度快、干燥效果好、智能程度高的脉动真空灭菌器进行无菌保障。本文以脉动真空湿热灭菌器验证为基础,根据制药生产企业和医疗单位对无菌保障的要求,开展多种不同物品同时用脉动真空灭菌器进行灭菌的效果验证,现报道如下。
1材料与方法
1.1材料和试剂
灭菌对象是疫苗生产中常用的硅胶管道、敷料包、橡胶栓、过滤器、超声破碎仪、玻璃吸管、无菌服、止血钳及器械等。采用XG1.DTF-1.5B型脉动真空灭菌器(山东新华医疗器械股份有限公司),设备自带温度/压力记录仪。后安装的SR10000型温度曲线自动记录仪(日本横河电机株式会社)。
灭菌验证效果所用的器材包括EB110型无线温度记录器(德国颐贝隆公司);Ebro温度验证记录仪(德国颐贝隆公司);Ebro压力验证记录仪(德国颐贝隆公司);132℃高压灭菌指示条和布维-狄克(B-D)真空试验测试图(山东新华医疗器械股份有限公司);自含菌为嗜热脂肪芽孢杆菌孢子(≥10-6)的脉冲指示管(美国MesalabsEZTest公司);实验用硫乙醇酸盐培养基(长春生物制品研究所有限公司,批号:20181205);DH3600BⅡ型35℃恒温培养箱(天津泰斯特仪器有限公司)。
1.2验证方法及评价标准
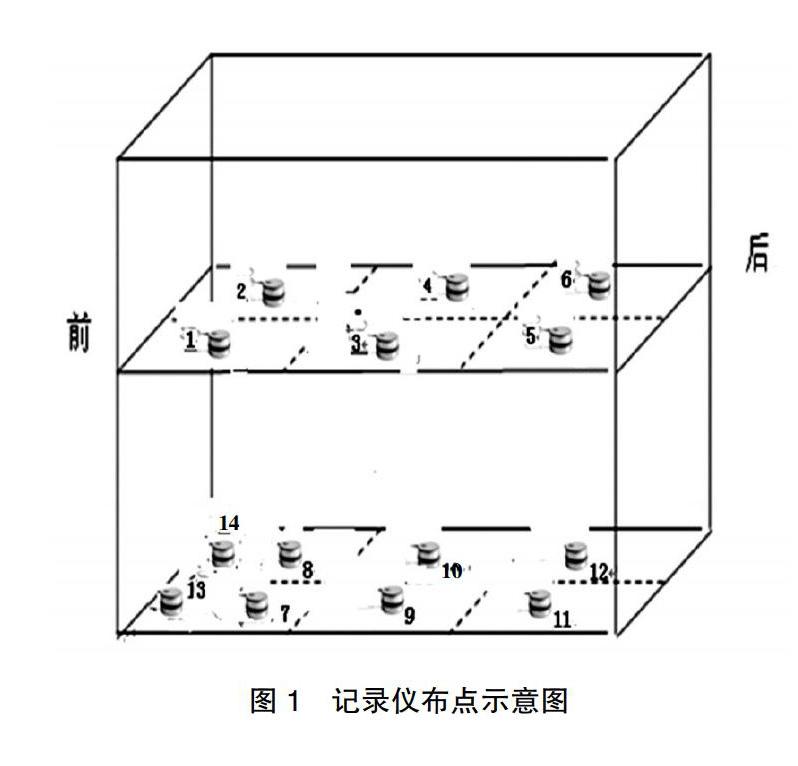
1.2.1灭菌器性能验证 具体操作步骤如下。①真空保压试验:确认空载状态下在灭菌器腔室内放置1个压力探头,启动脉动真空灭菌器保压程序,参数设定如下。保压限度-93 kPa,时间900 s,泄露限度≤2 kPa。结果判定,若900 s后初始压力≤-93 kPa、泄露率≤2 kPa/15 min,则视为设备保压合格。②B-D试验:灭菌器在无负载的初始状态下将B-D测试包置放于排气口上方20~30 cm处,并根据标记放置。设定B-D测试参数,脉动上下限±75 kPa,脉动3次,灭菌温度132℃,灭菌时间为4 min,在完成灭菌操作程序后,取出测试包并打开观察B-D指示图。结果判定,若B-D指示图上的色列条变化均匀,与对照图设计的颜色一致提示脉动真空灭菌器能够排除冷空气99%以上,灭菌器真空性能合格。③空载热分布试验:灭菌器在无负载的初始状态下,将经过校准合格并处在有效期内的14个Ebro自动温度、压力监控记录仪,以几何分布的方式放置在灭菌器的内室中,保证14个监控记录仪的每个位置都不小于距离灭菌器内室四壁5 cm的距离(图1)。在输水口处至少放置一个压力温度监控仪,并设定监控仪的读取记录频次为10 s/次。同时必须保证设备和14个监控记录仪同时开启和结束;选择132℃、8 min、脉动±75 kPa灭菌程序进行试验。灭菌操作完成后读取并记录每个点位的溫度数值。结果判定,若在灭菌过程中同一时间不同点位间的温度差别(均一性)≤2℃,且灭菌器主控温度探头与监控记录仪记录的温度数值差值均≤1℃,提示灭菌室内的温度分布可确定为均匀(但是在14个温度监控记录仪所监控的点位也有相对的冷点,将其标明,在下面的挑战试验中密切观察);否则视为有“最冷点”并判定此次空载试验失败。④满载热穿透试验:灭菌器装载不同的待灭菌物品必须按预先设计的规定位置摆放(此后也要严格执行),待达到满负荷装载后,将14个监控记录仪按空载试验放置同上,必须保证设备和14个监控记录仪同时开启和结束。选择132℃、8 min、脉动±75 kPa灭菌程序进行试验。灭菌程序结束后进行结果判定,若所有测试点标准灭菌时间(F0)≥15 min,提示灭菌器在规定的装载方式且满载的条件下满足灭菌要求。
1.2.2生物挑战试验验证 灭菌器装载材质、包装、体积、耐热系数等各不相同的待灭菌物品时必须按预先设计的规定位置摆放(此后也要严格执行),待达到满负荷装载后,将经过校准合格并处在有效期内的嗜热脂肪芽孢杆菌孢子按着满载热分布试验所测试的每个点位的不同物品中放置1份生物指示剂并在灭菌器外留有一份阳性对照。按照设定的灭菌程序(132℃、8 min、脉动±75 kPa)进行灭菌操作,结束后将经过挑战的生物指示剂和阳性对照同时在55~56℃下培养24 h。结果判定,经灭菌挑战后的嗜热脂肪芽孢杆菌孢子指示剂管内颜色呈现紫色,(说明书给定灭菌后的合格颜色)即为培养阴性(-),且阳性对照管内的生物指示剂颜色不变(黄色),可判定灭菌效果合格并达到灭菌验证要求同时需在质量控制(QC)实验室进行培养,证明无菌生长。若被挑战的生物指示剂有1支或多支指示剂的管内培养液仍为黄色,则判定灭菌效果验证失败。
1.2.3无菌试验 在灭菌效果验证合格的灭菌物品中,再分别抽取硅胶管道、敷料包、橡胶栓、过滤器、超声破碎仪、玻璃吸管、无菌服、止血钳及器械等样品在百级洁净环境下由QC人员用无菌水冲洗物品并收集淋洗水。采用硫乙醇酸盐培养基培养淋洗水的方法进行检测,在35℃培养箱中培养3 h。结果判定,所有接种培养基的淋洗水样品在3次培养中均无菌生长,则判定无菌验证通过,若有1例样品长菌则视为验证失败。
2结果
2.1保压试验结果
脉动真空灭菌器保压程序停止900 s后,初始压力仍≤-93 kPa、泄露率≤2 kPa,保压试验合格。
2.2 B-D试验结果
在空载条件下脉动上限+75 kPa、脉动下限-75 kPa、灭菌温度132℃、灭菌时间4 min、在完成灭菌操作后,取出测试包打开观察B-D指示图呈现色列条变化均匀与对照图设计的颜色一致,则脉动真空灭菌器能够排除冷空气99%以上,灭菌器的真空性能试验合格。
2.3空载热分布试验结果
在完成了一个灭菌周期(132℃、8 min、脉动±75 kPa)后读取监控记录仪。数据显示,在灭菌过程中的任何相同时间不同位置间的温度差别(均一性)均≤2℃,且灭菌器温度控制探头与监控记录仪记录的温度差值均≤1℃,空载热分布试验合格。
2.4满载热穿透试验结果
按着规定的装载方式且达到满负荷装载后选择132℃、8 min、脉动±75 kPa灭菌程序进行试验。灭菌程序结束后读取监控记录仪数据显示,所有的测试点F0≥15 min,满载热穿透试验合格。
2.5微生物挑战试验结果
芽孢指示剂按着满载热分布试验所测试的每个点位的不同物品中布放,完成1个灭菌程序操作后,取出所有被挑战的生物指示剂观察颜色并同阳性一起在55~56℃下培养24 h。被灭菌后的生物指示剂管内颜色呈现紫色,与说明书判定合格颜色一致,为培养阴性(-),同时55~56℃培养24 h后无细菌生长,微生物挑战试验合格。
2.6无菌试验结果
硅胶管道、敷料包、橡胶栓、过滤器、超声破碎仪、玻璃吸管、无菌服、止血钳及器械等物品的淋洗水在接种培养基中放置在35℃培养箱中培养3 h后,培养基中没有菌落生长,无菌试验合格。
3讨论
本试验首先通过了灭菌器的性能验证、微生物挑战试验验证和无菌试验验证,结合蒸汽灭菌的三大基本要素,即作用时间、灭菌温度和饱和蒸汽的质量[7],总结以上所有试验结果提示脉动真空灭菌器在选择132℃、8 min灭菌程序的条件下多种不同物品同载进行脉动真空灭菌验证合格,且符合现行GMP关于对灭菌法的要求。同时不仅证明了本台脉动真空灭菌器设备本身的可靠性,关键是对灭菌程序进行了验证,保证了灭菌效果的有效性[8]。同时密切观察在最冷点放置的生物指示剂是否通过微生物挑战性试验从而进一步证明灭菌的效果[9]。目前我国部分制药企业和医疗单位仍沿用121℃、30 min;121℃、15 min以及116℃、40 min的程序进行灭菌[10],存在灭菌品种单一、升温缓慢、灭菌时间相对较长、干燥效果不理想等情况。本研究根据制药企业和医疗行业中对灭菌物品多样化的具体特点进行分析,采用了多种物品同时用脉动真空灭菌器(对于非液体类的)进行132℃、8 min灭菌程序试验,试验结果显示,不仅安全性能得到保障,而且灭菌时间短、灭菌效果可靠、干燥效果好[11],可以在制药企业和及医疗行业中推广应用。
单一品种物品的灭菌无论从灭菌过程还是灭菌程序的验证都相对容易和简单,但实际工作情况并非如此,大多数行业都是不同物品同时进行灭菌,这样就要求必须要严格按着验证过程中的包装方式和装载位置进行包装和装载。以保证灭菌的有效性和均一性[12]。如果包装方式和装载位置发生改变必须重新做灭菌效果验证。如果遇到透气效果不好、耐热系数高的被灭菌物品可采用提高灭菌温度和延长灭菌时间的方法来解决,但无论采用何种灭菌参数,都必须验证所采用的灭菌工艺和监控措施。在日常的工作过程中能确保物品灭菌后的无菌保证水平(SAL)≤10-6。验证及操作人员应具备极强的责任心和专业理论水平[13]。
综上所述,灭菌效果验证是药品生产企业和医疗行业中质量安全的必要条件和保障,设备的验证和装载方法的验证要定期化和常态化并且符合设备验证的工艺要求[14],验证后在日常生产中的每次灭菌也必须对各项参数和Fo值进行监测[15]。也是对药品生产质量和医疗工作的重要保障。
[參考文献]
[1]纪邵梅,赵宏大.高压蒸汽灭菌器灭菌效果的验证方法[J].中国药事,2004,18(5):328.
[2]王苗,靳寸朵,赵小丽,等.高压蒸汽灭菌中湿包产生的原因及改进措施[J].实用临床护理学电子杂志,2018,3(10):191,195.
[3]焦家林,李敏.脉动真空灭菌器湿包常见原因及防范措施[J].医疗装备,2017,30(19):75.
[4]孔静.医疗器械的灭菌方法以及灭菌效果的验证[J].科技与企业,2012,21(4):223-224.
[5]过琴媛,李芙.疫苗生产工艺的验证[J].中国生物制品学杂志,2008,21(12):1135-1136.
[6]何国强.制药工艺验证实施手册[M].北京:化学工业出版社,2012.
[7]邓晓东,吕健,夏唯,等.医用纱布蒸汽灭菌验证[J].华南国防医学杂志,2014,28(7):704-707.
[8]国家药典委员会.中华人民共和国药典 [M].三部.北京:中国医药科技出版社,2015.
[9]张扬,张京.制药过程中高压蒸汽灭菌效果的验证[J].中日友好医院学报,2001,15(6):347-349.
[10]马延明,杨勇,王涛,等.无热源蒸汽灭菌在疫苗生产中的应用与效果验证[J].微生物学免疫学进展,2009,37(7):26-29.
[11]刘晓霞,闫虹.20家医疗机构压力蒸汽灭菌器灭菌效果检测分析[J].实用医技杂志,2018,25(7):748-749.
[12]王舰兵,李铎,徐康,等.132℃灭菌法在疫苗生产中的灭菌效果验证[J].中国消毒学杂志,2017,34(5):419-421.
[13]张小莉.药品微生物限度检查误差的影响因素及控制方法[J].中国药业杂志,2018,27(6):90-92.
[14]彭景峰.高压蒸汽灭菌设备的偏差管理与维修[J].设备管理与维修,2018,39(24):51-52.
[15]夏正明.蒸汽灭菌验证中的温度测量系统[J].上海医药,1997,19(2):33.
(收稿日期:2019-01-31 本文编辑:刘克明)
