不同配比青黛和雄黄对大鼠体内青黛指标性成分药代动力学研究
2019-10-28吴青青汪电雷黄和平
郭 艳, 吴青青, 汪电雷, 黄和平, 俞 娟
(安徽中医药大学 药学院,安徽 合肥 230012)
青黄散是由青黛和雄黄两味中药组成,以青黛为君药,具有凉血解毒、散瘀消积之功效,早在《世医得效方》和《奇效良方》中就有记载,属砷复合制剂[1]。方中青黛味咸、性寒,入肝经,能清热解毒,凉血消斑,泻火定惊,宜冲服或入丸散,其主要化学成分为靛蓝、靛玉红和色胺酮,具有抗菌消炎[2]、镇痛[3]、保肝[4]、抗肿瘤[5]、免疫调节等作用[6-7]。靛玉红是青黛抗白血病的主要药效成分,对慢性粒细胞白血病具有明显抑制作用[8]。雄黄辛温有毒,可解百毒,消积聚,化腹中瘀血[9-10],其主要成分二硫化二砷(As2S2)可抑制肿瘤细胞增殖,降低端粒酶活性,以诱导细胞凋亡[11-14]。雄黄外敷具有消炎、抗病毒等作用[15],内服具有抗风湿、抗肿瘤的作用,常用于急性髓系白血病在内的多种恶性血液病的治疗[16]。雄黄遇热易转化为有毒的As2O3,通过“水飞法”炮制雄黄以去除可溶性的As2O3,降低其毒性[17-18]。现代药理研究表明,青黛与雄黄伍用可显著增强其诱导白血病细胞凋亡的作用,常被用于血液疾病如白血病、骨髓增生异常综合征等的治疗[19-21]。研究发现,青黄散中青黛和雄黄不同剂量配比对体内砷的变化规律,结果表明青黄散中青黛比例的增加能促进大鼠体内砷的吸收[22]。为进一步考察青黛与雄黄配比变化对大鼠体内青黛指标性成分的药动学行为及青黛吸收的影响,本实验固定方中青黛的剂量,通过改变雄黄的剂量,采用UPLC-MS/MS法测定给药后血中青黛指标成分色胺酮、靛玉红和靛蓝的浓度并比较其药动学参数,深入探讨其在体内的相互作用关系,为其在临床应用提供理论依据。
1 材料与方法
1.1 实验仪器
Agilent 1290超高效液相色谱仪(美国Agilent公司);Triple Quad TM 4500 质谱仪(美国AB SCIEX公司);AS5150A型超声清洗仪(Autoscience公司);Dragonmed移液器(上海大龙医疗设备有限公司);BP211D型电子天平(德国Sartorius公司);Milli-Q超纯水器(美国 Millipore公司)。
1.2 供试材料
青黛(批号:170907)和雄黄(批号:171023)均购于安徽省亳州中药材市场,并经安徽中医药大学彭华胜教授鉴定为正品。标准品(纯度均≥98%):靛玉红(批号:110717)和靛蓝(批号:67707224)均购于中国食品药品检定研究院;色胺酮(韶远科技上海有限公司,批号:SY017117);卡马西平(成都德斯特生物技术有限公司,批号DST171120-318);N,N-二甲基甲酰胺(色谱纯,上海阿拉丁生化科技有限公司);乙腈(色谱纯,合肥科申化工有限公司);水为超纯水,其余试剂均为分析纯。
1.3 试验动物
雄性SD大鼠,体质量为180~220 g,由安徽医科大学实验动物中心提供,合格证号:SCXK(皖)2017-001,实验前禁食12 h,可自由饮水。
1.4 试验方法
1.4.1 色谱条件与质谱条件 Kinetex C18(100 mm×2.10 mm, 1.7 μm)色谱柱,流动相为乙腈-水(53∶47,V/V),流速为0.2 mL/min, 柱温为30 ℃,进样量为2 μL。采用电喷雾离子化(ESI)以多反应监测模式(MRM)进行正离子扫描。气路参数:喷雾电压(IS)为5.5 kV,离子化温度(TEM)为550 ℃;碰撞气(CAD)为10 psi、雾化气(GS1)为18 psi、加热气(GS2)为55 psi。用于定量分析的离子对和去簇电压(DP)及碰撞电压(CE)分别为:m/z 249.2→130.0,60 eV,38.63 eV(色胺酮);m/z 263.0→235,77.22 eV,34.3 eV(靛玉红);m/z 263.0→235,77.22 eV,34.3 eV (靛蓝);m/z 237.2→194.1,104.04 eV,27.8 eV (卡马西平)。
1.4.2 供试品的制备 称取1.0 g雄黄于研钵中,加0.5 mL水研磨5 min至糊状,再向研钵中加入40 mL水搅拌1 min,静置4 min,取混悬液,下沉的粗粉继续研磨,反复操作10次,合并混悬液,静置10 h以上,取下沉物冷冻干燥[23-24]。称取3份青黛粉末各10.0 g,按照190.0∶1、95.0∶1、47.5∶1的配比分别称取上述冷冻干燥后的雄黄52.6、105.3、210.5 mg,然后均以18 mL 0.5% CMC-Na和2 mL乙醇制成溶液,灌胃给药,给药体积为10 mL/kg。
1.4.3 对照品及内标溶液的配制 称取适量各对照品,分别用N,N-二甲基甲酰胺溶解,得到浓度均为100 μg/mL的各对照品储备液。分别移取不同体积各储备液于10 mL容量瓶中, 乙腈定容, 得色胺酮、靛玉红和靛蓝浓度分别为200、2 000和2 000 ng/mL的混合对照品储备液。精密称取卡马西平1.00 mg于10 mL容量瓶,乙腈溶解并定容,得浓度为100 μg/mL的内标储备液。取适量该储备液,用乙腈稀释成浓度为0.1 ng/mL的内标溶液,4 ℃下保存备用。
1.4.4 血浆样品处理 取血浆90 μL置于1.5 mL EP管中,加入10 μL内标和750 μL乙酸乙酯,涡旋3 min,12 000 r/min离心10 min,取上清液,氮吹,加50 μL乙腈复溶,涡旋混匀,再次12 000 r/min离心10 min,取上清液。
1.5 药代动力学实验
取18只SD大鼠,随机分为A、B、C三组,实验前禁食12 h,可自由饮水。以青黛剂量为5 g/kg,雄黄剂量分别为0.026 g/kg(A组)、0.053 g/kg(B组)、0.105 g/kg(C组)的配比进行大鼠灌胃给药,并于给药前和给药后5、15、30、60、120、240、360、480、600 min眼底静脉丛取血0.3 mL,置于经肝素处理的EP管中,3 500 r/min离心10 min,分离出血浆,按“1.4.4”节下血浆样品处理操作,进样分析。
1.6 数据处理
运用DAS 2.0软件进行药动学参数的计算。采用SPSS 23.0软件对数据进行分析,结果以“均值±标准差”表示,P<0.05表示差异显著。
2 结果与分析
2.1 专属性考察
分别取大鼠空白血浆、空白血浆加内标、空白血浆加内标和混合对照品溶液以及给药6 h后的大鼠血浆样品,按“1.4.4”节进样分析。如图1所示, 血浆中内源性物质对色胺酮、靛玉红、靛蓝和卡马西平的测定无干扰。

图1 大鼠空白血浆(A)、空白血浆加内标(B)、空白血浆加内标和混合对照品溶液(C)以及青黛∶雄黄=95∶1配伍给药6h后的大鼠血浆样品(D)的MRM色谱图,1.卡马西平;2.色胺酮;3.靛蓝;4.靛玉红Fig.1 MRM chromatograms of rat blank plasma (A), blank plasma plus internal standard (B), blank plasma plus internal standard and mixed reference solution (C), as well as rat plasma samples after 6 h of administration (D, indigo naturalis∶realgar=95∶1), 1. Carbamazepine; 2. Tryptanthrin; 3. Indigo; 4. Indirubin
2.2 标准曲线的绘制
用乙腈依次稀释混合对照品储备液,得到色胺酮浓度为2,4,10,20,50,100,200 ng/mL,靛玉红和靛蓝浓度分别为20,40,100,200,500,1 000,2 000 ng/mL的混合对照品溶液。取大鼠空白血浆90 μL,分别加入10 μL一系列混合工作液和750 μL乙酸乙酯,按“1.4.4”节下进样分析。以样品浓度为横坐标(x),以样品与内标物峰面积比值为纵坐标(y)进行线性回归,绘制标准曲线。得到方程:色胺酮:y=0.557x+0.327 4(r=0.996 1);靛玉红:y=0.010 3x-0.000 6(r=0.998 2);靛蓝:y=0.053 7x+0.229 6(r=0.997 3)。结果表明,色胺酮在0.2~20 ng/mL 浓度范围内线性关系良好,靛玉红和靛蓝在2~200 ng/mL浓度范围内线性关系均良好。
2.3 准确度考察
取大鼠空白血浆90 μL,加入一定量不同浓度的混合对照品溶液,分别配制成色胺酮、靛玉红和靛蓝定量下限(0.2,2,2 ng/mL)、低(0.4,4,4 ng/mL)、中(2,20,20 ng/mL)、高(10,100,100 ng/mL)浓度的质控样品,每个浓度平行5份,重复3个分析批,按“1.4.4”节下进样分析。结果如表1~3所示。
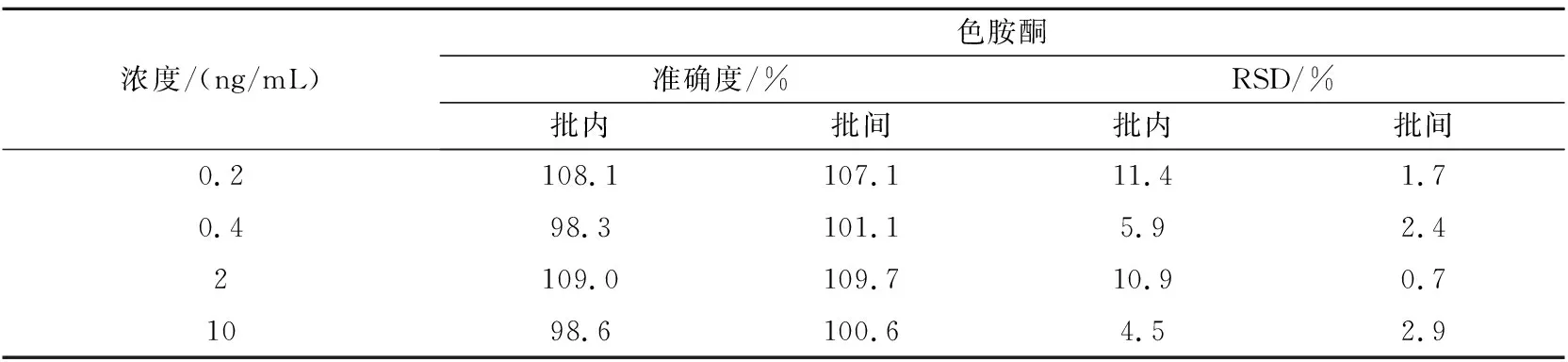
表1 色胺酮准确度测定(n=5)

表2 靛玉红准确度测定(n=5)
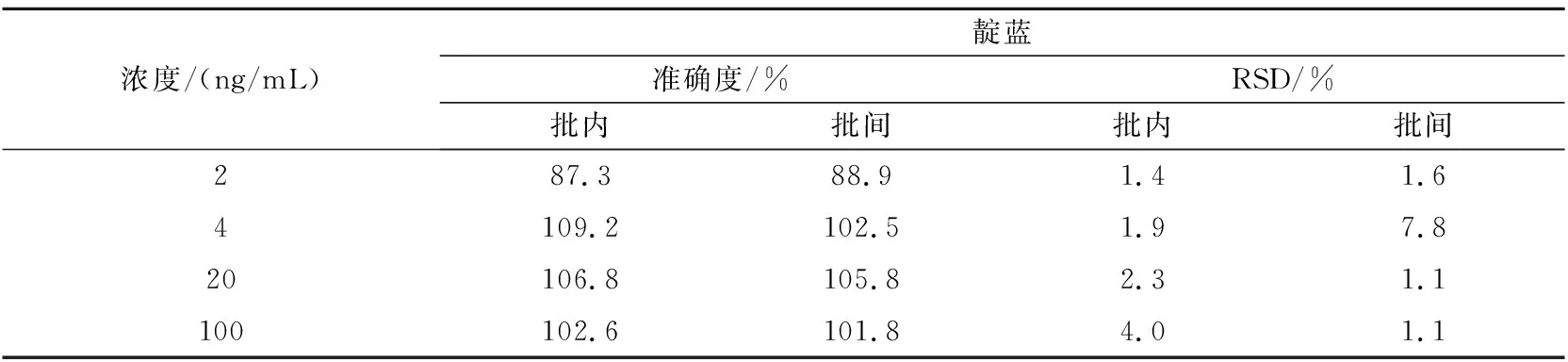
表3 靛蓝准确度测定(n=5)
2.4 基质效应
以大鼠空白血浆经乙酸乙酯处理后的空白基质上清液和乙腈为溶剂,分别配制含基质和不含基质的色胺酮、靛玉红和靛蓝低、中、高浓度的质控样品,每个浓度平行5份,进样分析。每个分析物及内标的基质因子为基质存在下的峰面积(由乙酸乙酯处理后的空白血浆加入分析物和内标测得)与不含基质的相应峰面积(分析物和内标的纯溶液)的比值。分析物与内标的基质因子的比值即为内标归一化的基质因子。结果表明内标归一化的基质因子的RSD均小于15%, 表明血浆基质对3种物质的测定影响较小。
2.5 提取回收率
以空白血浆90 μL,加入色胺酮、靛玉红和靛蓝低、中、高浓度的混合对照品溶液, 按血浆样品处理方法处理后的测定峰面积为A1,空白血浆样品按血浆样品处理方法处理后加入色胺酮、靛玉红和靛蓝低、中、高浓度的混合对照品测定的峰面积为A2,提取回收率=A1/A2×100%。结果表明低、中、高浓度下3种物质的提取回收率均大于85%,符合生物样品分析要求。
2.6 稳定性考察
配制色胺酮、靛玉红和靛蓝低、中、高浓度的质控样品,每个浓度平行5份。样品分别经反复冻融3次、室温放置6 h以及4 ℃放置24 h后, 按“1.4.4”节下进样分析。结果表明低、中、高浓度下3种物质的RE均小于15%,表明这3种物质在上述条件下均稳定。
2.7 青黛三种指标成分的血药浓度-时间曲线及其主要药代动力学参数
色胺酮、靛玉红和靛蓝的平均血药浓度-时间曲线如图2~4所示。采用DAS 2.0软件进行分析,得到的主要药动学参数结果见表4~6。

图2 色胺酮平均血药浓度-时间曲线(n=6)Fig.2 Mean plasma concentration-time curve of tryptanthrin
图3 靛玉红平均血药浓度-时间曲线(n=6)Fig.3 Mean plasma concentration-time curve of indirubin
图4 靛蓝平均血药浓度-时间曲线(n=6)Fig.4 Mean plasma concentration-time curve of indigo
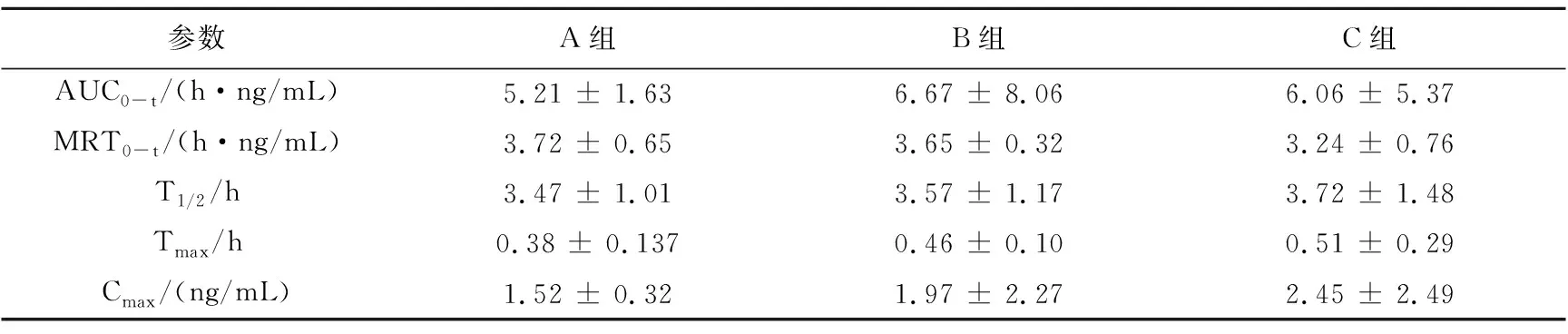
参数A组B组C组 AUC0-t/(h·ng/mL)5.21 ± 1.636.67 ± 8.066.06 ± 5.37 MRT0-t/(h·ng/mL)3.72 ± 0.653.65 ± 0.323.24 ± 0.76 T1/2/h3.47 ± 1.013.57 ± 1.173.72 ± 1.48 Tmax/h0.38 ± 0.1370.46 ± 0.100.51 ± 0.29 Cmax/(ng/mL)1.52 ± 0.321.97 ± 2.272.45 ± 2.49
表5 大鼠灌胃给药后靛玉红主要药动学参数
注:C组与A组比较,*P< 0.05;C组与B组比较,#P< 0.05。下同。
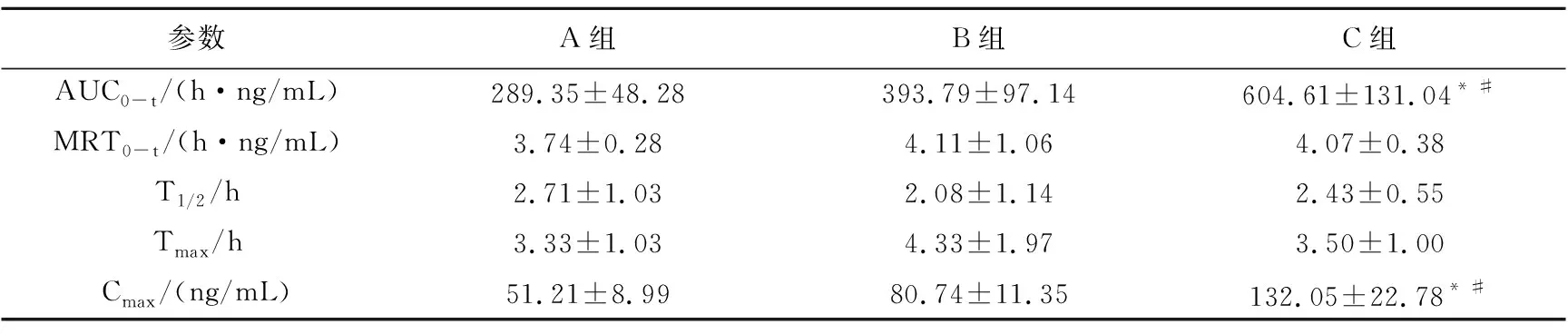
表6 大鼠灌胃给药后靛蓝主要药动学参数
3 结论与讨论
前期研究比较了甲醇、乙腈和N,N-二甲基甲酰胺等溶剂超声溶解效果,结果显示色胺酮、靛玉红及靛蓝在N,N-二甲基甲酰胺中均能较好地溶解,故实验选用N,N-二甲基甲酰胺溶解这三种物质。卡马西平以及各对照品在相同色谱条件下出峰位置合适,且无组分峰影响,实验选用卡马西平为内标物。青黛难溶于水,实验采用0.5%CMC-Na和少量乙醇溶解青黛粉末以制成混悬液灌胃给药。关于生物样品前处理,用乙酸乙酯萃取青黛中的靛玉红,乙腈沉淀蛋白效果好,氮吹效率高[23]。本实验还考察了甲醇-水、乙腈-水的不同比例流动相,结果表明以乙腈-水(53∶47,V/V)作为流动相时,待测物响应值较高,分离效果较好,因而实验选用乙腈-水(53∶47,V/V)为流动相进行指标成分浓度的测定。
一般来说,中药复方不同配比的选择可直接影响其临床疗效与安全[24]。青黄散中随着雄黄剂量的增大,其治疗白血病的效果增加,但雄黄剂量过大会造成体内砷蓄积而引起一系列毒副作用[25]。青黄散中青黛与雄黄常用比例为7∶3、8∶2、9∶1,但可能由于仪器等实验条件受限,前期预实验结果发现,青黛在较低剂量时很难同时测定青黛中三种指标成分的体内浓度。本实验在前人工作的基础上,通过比例调整,分别以青黛与雄黄比例为190.0∶1、95.0∶1和47.5∶1进行大鼠灌胃给药,测定给药后血中青黛指标成分的浓度并比较其药动学参数,探讨不同配比的青黛与雄黄伍用对体内青黛吸收的影响。结果显示,随着雄黄剂量的增大,色胺酮的Cmax、AUC0-t和MRT均无显著变化(P>0.05),说明雄黄剂量变化不影响色胺酮的吸收;靛蓝的Cmax和AUC0-t均显著增大(P<0.05),说明雄黄剂量增加能促进青黛中靛蓝的吸收[22];靛玉红的Cmax和AUC0-t均减小(P<0.05),可能是由于青黛中的靛玉红与雄黄中的砷化物相互作用形成有机配合物发挥其抗肿瘤作用而使血中靛玉红浓度降低,这可能是青黛与雄黄伍用治疗白血病效果更优的一个原因[26]。综上可知,不同配比的青黛与雄黄伍用时,青黛的体内药动学行为发生改变,雄黄剂量增大能促进青黛吸收,但不是促进全部有效成分吸收。而对于青黛与雄黄伍用治疗血液疾病的确切作用机制还有待进一步研究。
